(I notice that my overall page views seem to have dropped off quite a bit since the test yesterday. Good job, guys.)
(EDIT 17/6/16: I noticed that there were some errors with regards to the half-life equations. I have fixed them up, and hopefully they should be correct now.)
1) Show awareness of how plasma drug concentration versus
time curves provide valuable insight into the pharmacokinetic
properties of any drug
Plasma drug concentration versus time curves are a great way to show trends in plasma concentrations of drugs (well, that's pretty much what they're meant to do, by definition). Also, if you calculate the area under the curve, by integration or otherwise, you can find out the patient's total exposure to a drug over a given time. A larger area under a curve would suggest that the drug is better absorbed. As well as this, you can also use the area under a curve to calculate other pharmacokinetic parameters, such as clearance: more on this in a bit.
2) Show an understanding of the concept of “clearance” and
how it is determined for a given drug
Clearance is the volume of blood cleared of the drug per unit of time (and is hence measured in units such as L/hr or mL/min). It is also the constant that relates the plasma drug concentration with the rate of elimination. "Total body clearance" refers to the overall clearance, whereas "renal clearance" and "hepatic clearance" refer to clearance by the kidneys and liver, respectively. To determine the clearance, you first need to give the patient a single IV dose of the drug. (Oral won't work, because there are too many confounding factors surrounding the absorption of oral drugs.) After that, you need to take blood samples at various time points and work out the plasma drug concentration at each time point. These points can then be plotted onto a curve and the area under the curve calculated. Finally, to work out the clearance, simply divide the dose by the area under the curve.
Clearance is important in determining drug concentration at "steady state." Steady state is the state in which the rate of drug administration is equal to the rate of drug elimination.
Now, as I've mentioned before, clearance is the constant that relates the plasma drug concentration with the rate of elimination. Hence:
Clearance * Plasma drug concentration = elimination rate
However, when the plasma drug concentration = steady state drug concentration, elimination rate = rate of drug administration. Hence:
Clearance * Steady state drug concentration = Rate of drug administration
Again, this gets a bit more complicated during oral dosing. There isn't really a "steady state drug concentration" in oral dosing as the drug isn't being constantly infused- rather, it is taken over several intervals. However, to my understanding, this equation is still somewhat applicable, but you have to replace "steady state drug concentration" with "average drug plasma concentration between dosing intervals," which is a bit of a mouthful.
3) Show an appreciation of the “volume of distribution” and how
it is estimated together with an awareness of how this value
reveals the behaviour of drugs within the human body
I feel like I've already spoken about this before. Oh wait, I have, on an earlier post: Drug Absorption and Distribution.
Now I'm going to go a bit further and talk about how the volume of distribution is estimated. It's best estimated using the plasma drug concentration at zero time, as you know that no drug could have been metabolised by that point. Once again, IV dosing is used so as to avoid the confounding variables of absorption by the gut and so forth. Blood samples are collected at various intervals, and then the curve is extrapolated back to find the plasma drug concentration at t = 0. The original dose is then divided by this plasma drug concentration to give the volume of distribution.
Why is the volume of distribution important? Volume of distribution can be useful for helping us determine how to achieve a therapeutic drug concentration in a short period of time. As the volume of distribution = (dose)/(plasma drug concentration), then the dose required to reach a particular plasma concentration can be calculated by dose = (volume of distribution)*(plasma drug concentration).
4) Be able to provide a simple sketch to show an appreciation of
the concept of drug “half-life” together with an awareness of
how it is estimated in human subjects.
Okay well screw the "simple sketch" part because I'm too lazy to draw a diagram. Half-life is probably a concept that you've encountered before, though: it's simply the time that it takes for the plasma concentration of a drug to drop by 50%. Half-life is not considered to be a fundamental pharmacokinetic parameter (as opposed to clearance and volume of distribution) as it is determined by clearance and volume of distribution.
Plasma drug concentrations can also be described using a nice little exponential equation:
Ct = C0 e^(-kt)
where Ct = concentration at time t, C0 = concentration at time 0, k = the elimination rate constant (which is the proportion of drug removed in an hour, or whatever time units you're using) and t = time.
This can then be used to determine half-life. You see, after the first half life, Ct = 0.5C0. Hence the equation can be rearranged to directly link k and t:
0.5C0 = C0 e^(-kt)
0.5 = e^(-kt)
ln 0.5 = -kt
-k = (ln 0.5)/t
-k = (ln 2^(-1))/t
-k = (-ln 2)/t
k = (ln 2)/t
You may also see this equation written as k = 0.693/t. It's the same thing really: 0.693 is the natural log of 2.
As I mentioned before, half-life is determined by clearance and volume of distribution. Naturally, there's an equation linking these three variables:
t = (0.693*V)/CL (where t = time at the first half-life).
Hence, half-life is increased by an increased volume of distribution, but decreased with an increased clearance. This makes sense: the more of the drug that's "filling up" your body, the more time you'll need to get rid of it. Also, if clearance (which is constant for a particular drug and a particular patient) is high, then it'll be cleared pretty quickly and so half-life will be low.
5) Define the concept of “oral bioavailability,” showing a basic
awareness of the factors that influence it together with how it
is determined experimentally.
Bioavailability, sometimes denoted by the letter F (presumably B was already taken up, or "bioavailability" starts with the letter F in some weird language), is essentially the proportion of drug that reaches the systemic circulation. IV drugs pretty much all have a bioavailability of 1 as they are taken directly into the bloodstream. Oral drugs are a bit different, however: they must first be absorbed by the gut, and then passed through the liver. Not all of a drug will make it through the liver, as some of it will be metabolised there. The overall bioavailability for an oral drug can be calculated by multiplying the percentage that was absorbed via the gut by the percentage that makes it through the liver in its original form.
There are other ways of calculating bioavailability, again using the area under a curve. Bioavailability can be calculated by dividing the area under a curve for the oral dose by the area under a curve for the IV dose. If the oral and IV doses are different, just use this nifty formula:
F = (AUC(oral)*DOSE(IV))/(AUC(IV)*DOSE(oral))
where F = bioavailability and AUC = area under the curve
Aaaaaaaaaaand I think that's pretty much it for this lecture! (At some point I need to revise my Research and Communications Exercise, though. I wrote a helluva lot of bullshit on steady state concentrations and stuff in there, and it's probably not very accurate. This unit coordinator is pretty merciless- and he's going to be the one marking the assignment. Ah well, I still have over a month to fix it up :) )
Thursday, March 31, 2016
Tuesday, March 29, 2016
Intrinsic Postvertebral Muscles
Okay, I should probably stop soon and focus on cramming for those embryo slides, since that's what the test is going to be focused on. But oh well.
Vertebral Ligaments
First, I'm going to touch on the vertebral ligaments. I've touched on the annulus fibrosus, the outer layers of which join the endplates of adjacent vertebrae together. Here's a list of some others:
The three unisegmental muscles are, as their name suggests, muscles that only run between one segment. The intertransverse muscle pretty much runs along with the intertransverse ligament and the interspinous muscle pretty much runs along with the interspinous ligament. The third unisegmental muscle, the levatores costarum, lifts the ribs.
Transversospinales
Transversospinales are muscles that run from a transverse process UP to a spinous process of a vertebra above it. There are three main types.
Rotatores muscles, found only in the thoracic region, run from a transverse process to the spinous process of a vertebra 1 or 2 segments above the first. Their fibres are almost horizontal. As their name suggests, they are good at rotating things.
Multifidus muscles are found everywhere in the vertebral column, but are less important in the thorax. They cross 3 or 4 segments.
Finally, semispinalis muscles, found only in the upper back, cross 5 or 6 segments.
Erector spinae
The erector spinae muscles run longitudinally up the spine. They all begin inferiorly as the sacrospinalis tendon, and each fibre runs 6 segments before "passing the baton" to another fibre which runs up another 6 segments. Spinalis muscles attach to the spinous processes, longissimus muscles attach to the transverse processes and iliocostalis muscles attach to the ribs (costalis = ribs).
Before I move on, I'm just going to quickly mention a few more points. All of the muscles that I have spoken about so far are known as the intrinsic postvertebral muscles. They are all derived from the epimere, and therefore their nerve supply is from the dorsal rami.
There is one more muscle that is derived from the epimere. It's not part of the erector spinae, but I don't know where else to put it so I'm going to put it here. It's called the splenius. The splenius, in contrast to the transversospinales, is a spinotransverse muscle. That means that it runs from the spinous process of a vertebra UP to the transverse processes. They arise from the upper thoracic and cervical spinous processes. The splenius cervicis, which is the lower part, inserts on the transverse processes of C1 to C4. The splenius capitus, which is the upper part, goes up to the nuchal line of the skull.
Other muscles derived from hypomere
Aside from the above muscles derived from the epimere, there are several muscles derived from the hypomere. Some of the deeper ones include the serratus posterior superior and serratus posterior inferior. These go from the upper 4 or lower 4 thoracic spinous processes to the upper 4 or lower 4 ribs, respectively. Of course, there are many other muscles involved, like the obliques and stuff, but I won't go into those now.
The thoracolumbar fascia
The thoracolumbar fascia is a sheet of connective tissue that encloses the intrinsic postvertebral muscles in the thoracic and lumbar regions (hence "thoraco" and "lumbar"). In the thorax, the fascia is thin and stretchy to accommodate breathing, but in the lumbar region it's tougher (in order to provide stability) and has three layers. The posterior layer attaches to the spinous processes, the middle layer attaches to the transverse processes and the anterior layer attaches to the vertebral bodies. The three layers all meet laterally to the vertebrae, and then continue on to form the transversus abdominus, which is one of the muscles of the abdominal wall. The muscles I have talked about so far (well, aside from the serratus posterior) are enclosed within the middle and posterior layers. Between the anterior and middle layers lie two more muscles: the anterior one is the psoas, whereas the posterior one is the quadratus lumborum.
Aaaaand now I should take a break from stressing about muscles! (Now I'm just going to stress about something else instead... bugger.)
Vertebral Ligaments
First, I'm going to touch on the vertebral ligaments. I've touched on the annulus fibrosus, the outer layers of which join the endplates of adjacent vertebrae together. Here's a list of some others:
- Anterior longitudinal ligament- runs all the way up the anterior side of the vertebral bodies. Multisegmental (i.e. crosses many segments).
- Posterior longitudinal ligament- runs all the way up the posterior side of the vertebral bodies. Multisegmental.
- Ligamentum flavum- joins the laminae of adjacent vertebrae. "Flavum" means "yellow," and it refers to the yellow-looking elastic tissue that makes up this ligament. The elasticity means that it doesn't buckle into the spinal cord, which is pretty important.
- Intertransverse ligament- connects the transverse processes of adjacent vertebrae.
- Capsular ligament of zygapophysial joints- surrounds the joints I guess?
- Interspinous ligament- joins the spinous processes of adjacent vertebrae.
- Supraspinous ligament- runs along the tips of the vertebrae. Multisegmental.
The three unisegmental muscles are, as their name suggests, muscles that only run between one segment. The intertransverse muscle pretty much runs along with the intertransverse ligament and the interspinous muscle pretty much runs along with the interspinous ligament. The third unisegmental muscle, the levatores costarum, lifts the ribs.
Transversospinales
Transversospinales are muscles that run from a transverse process UP to a spinous process of a vertebra above it. There are three main types.
Rotatores muscles, found only in the thoracic region, run from a transverse process to the spinous process of a vertebra 1 or 2 segments above the first. Their fibres are almost horizontal. As their name suggests, they are good at rotating things.
Multifidus muscles are found everywhere in the vertebral column, but are less important in the thorax. They cross 3 or 4 segments.
Finally, semispinalis muscles, found only in the upper back, cross 5 or 6 segments.
Erector spinae
The erector spinae muscles run longitudinally up the spine. They all begin inferiorly as the sacrospinalis tendon, and each fibre runs 6 segments before "passing the baton" to another fibre which runs up another 6 segments. Spinalis muscles attach to the spinous processes, longissimus muscles attach to the transverse processes and iliocostalis muscles attach to the ribs (costalis = ribs).
Before I move on, I'm just going to quickly mention a few more points. All of the muscles that I have spoken about so far are known as the intrinsic postvertebral muscles. They are all derived from the epimere, and therefore their nerve supply is from the dorsal rami.
There is one more muscle that is derived from the epimere. It's not part of the erector spinae, but I don't know where else to put it so I'm going to put it here. It's called the splenius. The splenius, in contrast to the transversospinales, is a spinotransverse muscle. That means that it runs from the spinous process of a vertebra UP to the transverse processes. They arise from the upper thoracic and cervical spinous processes. The splenius cervicis, which is the lower part, inserts on the transverse processes of C1 to C4. The splenius capitus, which is the upper part, goes up to the nuchal line of the skull.
Other muscles derived from hypomere
Aside from the above muscles derived from the epimere, there are several muscles derived from the hypomere. Some of the deeper ones include the serratus posterior superior and serratus posterior inferior. These go from the upper 4 or lower 4 thoracic spinous processes to the upper 4 or lower 4 ribs, respectively. Of course, there are many other muscles involved, like the obliques and stuff, but I won't go into those now.
The thoracolumbar fascia
The thoracolumbar fascia is a sheet of connective tissue that encloses the intrinsic postvertebral muscles in the thoracic and lumbar regions (hence "thoraco" and "lumbar"). In the thorax, the fascia is thin and stretchy to accommodate breathing, but in the lumbar region it's tougher (in order to provide stability) and has three layers. The posterior layer attaches to the spinous processes, the middle layer attaches to the transverse processes and the anterior layer attaches to the vertebral bodies. The three layers all meet laterally to the vertebrae, and then continue on to form the transversus abdominus, which is one of the muscles of the abdominal wall. The muscles I have talked about so far (well, aside from the serratus posterior) are enclosed within the middle and posterior layers. Between the anterior and middle layers lie two more muscles: the anterior one is the psoas, whereas the posterior one is the quadratus lumborum.
Aaaaand now I should take a break from stressing about muscles! (Now I'm just going to stress about something else instead... bugger.)
Regions of the Vertebral Column
Now the study's getting a bit more panicky, because the test is tomorrow :( On the upside, they did say that we'd only be tested on limited vertebral column stuff. On the downside, EMBRYO SLIDES. Ugh.
Evolution of regions
To examine the evolution of different regions of the spine, we're going to have a look at living creatures.
First we'll start off with fish. Fish have thoracic vertebrae (and therefore ribs), as well as caudal vertebrae (vertebrae past the anus). That's it. They don't have necks, because they can just swim onto their food.
Next we'll look at amphibians, because they spend some time in the water (like fish) and some time on the land (unlike fish). Amphibians also have thoracic and caudal vertebrae, but they also have sacral vertebrae because their hindlimbs need a firm attachment. And yes, they still have no neck. Instead, they have other specialisations for gaining food (maybe this is why frogs have long tongues? Who knows).
Reptiles are the next group of interest. They're a bit like amphibians, but they spend more time on the land. This is the first group to have a neck! Yay! This cervical (neck) region seems pretty important for land animals, but not for aquatic animals. Mammals that have returned to the sea, such as dolphins and whales, retain their cervical vertebrae, but they're flattened and quite rudimentary. Although reptiles are more advanced with their fancy-pants necks and all, they still move a lot like fish, with side-to-side undulations of their bodies.
Movement starts to become more developed with the addition of the lumbar region in mammals. This is kinda important because the alignment of the limbs of mammals (rotated under the body) isn't very conducive to the whole side-to-side undulations thing. Instead their lumbar regions allow them to have flex-extend locomotion, which allows for galloping and so forth. Mammals that return to the sea also have lumbar vertebrae, and also move by flex-extend locomotion.
The relative size of the thoracic and lumbar regions can also give a clue as to the preferred locomotion of the animal. You see, while the lumbar region is good for flexion and extension, the thoracic region is for axial rotation. Hence, terrestrial animals that need to run and gallop a lot have a more developed lumbar region, whereas arboreal (i.e. tree-dwelling) animals have a more developed thoracic spine, allowing them to swing from branch to branch.
Oh, and one last quick note before moving on. Birds have all of the main spinal regions (cervical, thoracic, lumbar, sacral and caudal). However, all of these regions, with the exception of the neck, are fused to some degree.
The motion segment
The motion segment, to my understanding, is a collective name for all the bits and pieces in the spine that allow for motion. Another way of looking at it is that the motion segment refers to two vertebrae plus the joints in between. The motion segment can be divided up into two sections: anterior elements and posterior elements. Anterior elements are more load-bearing (carrying 80% of the weight) and are comprised of the intervertebral discs and the vertebral bodies themselves. Posterior elements, on the other hand, are more responsible for the movement. Posterior elements include the bony bits (spinous processes, transverse processes and laminae) along with associated ligaments and zygapophyseal joints. (Yup, that's a longish word- I'll cover more on this later, I promise.)
The Intervertebral Disc (IVD)
In my previous post, I alluded to the development of the intervertebral disc. Time to go back into that topic with more gusto!
So, a recap plus a bit of expansion on what I talked about before. In my previous post, I mentioned that the notochord gets "pinched off" into the region between somites. The significance of this is that they begin to form the nucleus pulposus, which makes up the middle of the intervertebral discs. The notochordal cells themselves begin to degenerate at around 6 months gestation as they are replaced by acellular material derived from the annulus fibrosus (yes, yes, I'll get to that later), but some cells do remain until adulthood. The nucleus pulposus is eventually made up of a hydrophilic mixture made up of collagen fibrils, hyaluronic acid and proteoglycans, allowing it to retain water easily. (This water can be squeezed out during our everyday activities, which is why we're generally slightly shorter at night than in the morning.) As we age, the nucleus is gradually replaced with fibrous tissue and contains less water.
Alright, so you might be wondering what the annulus fibrosus is. It is made up of many layers of fibrocartilage that run in rings around the nucleus pulposus. Well, actually, pretty much only the inner layers run in rings around the nucleus pulposus- the outer layers just join the two endplates of adjacent vertebrae to each other.
This overall structure of a hydrophilic core surrounded by fibrocartilage is quite useful. Intervertebral discs are capable of transmitting loads and allowing movement. The "envelope" formed by the annulus fibrosus provides tensile strength which maintains the pressure in the nucleus pulposus, keeps the vertebrae apart and allows the vertebrae to move. However, this can go wrong: if the envelope is weak in any way, the nucleus can escape or the annulus can bulge and press on other structures.
Control of movement – Zygapophysial joints
The zygapophysial joints exist mainly between the inferior articular facet of one vertebra and the superior articular facet of the next. The angles of motion available depends to a large extent on the angles of the facets. In the thoracic region, the facets are set on the arc of a circle- very conducive to rotation. In the lumbar region, however, the facets are "radial" which means that rotation is not possible. Cervical region facets are oblique which allows for a wider range of movement.
The intervertebral discs also play roles in movement. In the lumbar and thoracic vertebrae, the joints are perpendicular to the plane of the disc so that some rotation occurs in the disc. In the cervical vertebrae, however, some translation occurs instead.
Regions and Ribs
Finally, I'm going to give a quick guide to how to tell the different vertebral regions apart.
Cervical vertebrae are somewhat easy, as they have transverse foraminae (i.e. little holes in the transverse processes).
Thoracic vertebrae can either be easy or hard to tell apart, depending on what you're provided with. Thoracic vertebrae are attached to ribs, so if there are ribs present, it's really easy. However, if there aren't, then you have to look for the rib facets (little bumps where the ribs attach). Sometimes the rib facets are really subtle and hard to see. Also, one point of interest is that most thoracic vertebrae actually have two demifacets- one at the top and one at the bottom. (Most ribs attach to two demifacets from adjacent vertebrae.) The exception is T12 which only has one rib facet.
I'm going to skip lumbar and go straight to sacral. Sacral are really easy, as they're all fused together.
Finally, lumbar is basically the "none of the above" category.
Evolution of regions
To examine the evolution of different regions of the spine, we're going to have a look at living creatures.
First we'll start off with fish. Fish have thoracic vertebrae (and therefore ribs), as well as caudal vertebrae (vertebrae past the anus). That's it. They don't have necks, because they can just swim onto their food.
Next we'll look at amphibians, because they spend some time in the water (like fish) and some time on the land (unlike fish). Amphibians also have thoracic and caudal vertebrae, but they also have sacral vertebrae because their hindlimbs need a firm attachment. And yes, they still have no neck. Instead, they have other specialisations for gaining food (maybe this is why frogs have long tongues? Who knows).
Reptiles are the next group of interest. They're a bit like amphibians, but they spend more time on the land. This is the first group to have a neck! Yay! This cervical (neck) region seems pretty important for land animals, but not for aquatic animals. Mammals that have returned to the sea, such as dolphins and whales, retain their cervical vertebrae, but they're flattened and quite rudimentary. Although reptiles are more advanced with their fancy-pants necks and all, they still move a lot like fish, with side-to-side undulations of their bodies.
Movement starts to become more developed with the addition of the lumbar region in mammals. This is kinda important because the alignment of the limbs of mammals (rotated under the body) isn't very conducive to the whole side-to-side undulations thing. Instead their lumbar regions allow them to have flex-extend locomotion, which allows for galloping and so forth. Mammals that return to the sea also have lumbar vertebrae, and also move by flex-extend locomotion.
The relative size of the thoracic and lumbar regions can also give a clue as to the preferred locomotion of the animal. You see, while the lumbar region is good for flexion and extension, the thoracic region is for axial rotation. Hence, terrestrial animals that need to run and gallop a lot have a more developed lumbar region, whereas arboreal (i.e. tree-dwelling) animals have a more developed thoracic spine, allowing them to swing from branch to branch.
Oh, and one last quick note before moving on. Birds have all of the main spinal regions (cervical, thoracic, lumbar, sacral and caudal). However, all of these regions, with the exception of the neck, are fused to some degree.
The motion segment
The motion segment, to my understanding, is a collective name for all the bits and pieces in the spine that allow for motion. Another way of looking at it is that the motion segment refers to two vertebrae plus the joints in between. The motion segment can be divided up into two sections: anterior elements and posterior elements. Anterior elements are more load-bearing (carrying 80% of the weight) and are comprised of the intervertebral discs and the vertebral bodies themselves. Posterior elements, on the other hand, are more responsible for the movement. Posterior elements include the bony bits (spinous processes, transverse processes and laminae) along with associated ligaments and zygapophyseal joints. (Yup, that's a longish word- I'll cover more on this later, I promise.)
The Intervertebral Disc (IVD)
In my previous post, I alluded to the development of the intervertebral disc. Time to go back into that topic with more gusto!
So, a recap plus a bit of expansion on what I talked about before. In my previous post, I mentioned that the notochord gets "pinched off" into the region between somites. The significance of this is that they begin to form the nucleus pulposus, which makes up the middle of the intervertebral discs. The notochordal cells themselves begin to degenerate at around 6 months gestation as they are replaced by acellular material derived from the annulus fibrosus (yes, yes, I'll get to that later), but some cells do remain until adulthood. The nucleus pulposus is eventually made up of a hydrophilic mixture made up of collagen fibrils, hyaluronic acid and proteoglycans, allowing it to retain water easily. (This water can be squeezed out during our everyday activities, which is why we're generally slightly shorter at night than in the morning.) As we age, the nucleus is gradually replaced with fibrous tissue and contains less water.
Alright, so you might be wondering what the annulus fibrosus is. It is made up of many layers of fibrocartilage that run in rings around the nucleus pulposus. Well, actually, pretty much only the inner layers run in rings around the nucleus pulposus- the outer layers just join the two endplates of adjacent vertebrae to each other.
This overall structure of a hydrophilic core surrounded by fibrocartilage is quite useful. Intervertebral discs are capable of transmitting loads and allowing movement. The "envelope" formed by the annulus fibrosus provides tensile strength which maintains the pressure in the nucleus pulposus, keeps the vertebrae apart and allows the vertebrae to move. However, this can go wrong: if the envelope is weak in any way, the nucleus can escape or the annulus can bulge and press on other structures.
Control of movement – Zygapophysial joints
The zygapophysial joints exist mainly between the inferior articular facet of one vertebra and the superior articular facet of the next. The angles of motion available depends to a large extent on the angles of the facets. In the thoracic region, the facets are set on the arc of a circle- very conducive to rotation. In the lumbar region, however, the facets are "radial" which means that rotation is not possible. Cervical region facets are oblique which allows for a wider range of movement.
The intervertebral discs also play roles in movement. In the lumbar and thoracic vertebrae, the joints are perpendicular to the plane of the disc so that some rotation occurs in the disc. In the cervical vertebrae, however, some translation occurs instead.
Regions and Ribs
Finally, I'm going to give a quick guide to how to tell the different vertebral regions apart.
Cervical vertebrae are somewhat easy, as they have transverse foraminae (i.e. little holes in the transverse processes).
Thoracic vertebrae can either be easy or hard to tell apart, depending on what you're provided with. Thoracic vertebrae are attached to ribs, so if there are ribs present, it's really easy. However, if there aren't, then you have to look for the rib facets (little bumps where the ribs attach). Sometimes the rib facets are really subtle and hard to see. Also, one point of interest is that most thoracic vertebrae actually have two demifacets- one at the top and one at the bottom. (Most ribs attach to two demifacets from adjacent vertebrae.) The exception is T12 which only has one rib facet.
I'm going to skip lumbar and go straight to sacral. Sacral are really easy, as they're all fused together.
Finally, lumbar is basically the "none of the above" category.
Monday, March 28, 2016
Drug Excretion
Back to studying Pharmacology!
So far, we've covered Absorption, Distribution and Metabolism- the first 3 letters of that ADME acronym that I've shown you before. Now I'm going to cover the final letter- Excretion!
1) Name and define the physiological processes that permanently rid the body of drugs and foreign chemicals.
Okay so I think this is just the definitions bit. There's a few important definitions that you have to know:
Excretion is the irreversible removal of the parent drug from the body. I *think* metabolism counts in this process, as the parent drug is being transformed into something that is... well, not the parent drug any more.
Elimination is the irreversible removal of the parent drug PLUS METABOLITES.
Clearance is the rate at which excretion occurs. (I think it's excretion rather than elimination anyway, as I'm fairly sure that the liver processing stuff is known as "hepatic clearance.") It is the volume of blood which is totally cleared of the drug per unit time and is thus measured in volume per time, such as litres per hour or millilitres per minute.
(Actually, I might need to double check some of these definitions. I'm currently asking a question on the PHAR2210 forums. I'll update this when I receive an answer- if I remember, anyway.)
One important acronym is fe: fraction excreted unchanged. It is the fraction of the drug that you pee out again in its original, unmetabolised form. (Sorry, I just put this here because I didn't know where else to put it.)
2) Identify organs and tissues playing the greatest role in removing drugs from the body.
The two main organs for removing drugs from the body are the liver and kidney. Other organs, such as the skin, lungs, hair and breasts (in lactating women) also play roles. I'll be covering all of these in greater detail throughout this post.
3) Identify three processes that control the excretion of drugs by the kidney.
The kidneys are a very important organ for removing drugs and their metabolites. Their functional units are called nephrons. Each nephron contains a glomerular capsule, which is where filtration takes place; proximal tubules, in which active secretion takes place; and the loop of Henle and distal tubules, where most reabsorption takes place.
The first process that occurs is filtration. The glomerular capsule, or Bowman's capsule, located at one "end" of the nephron, consists of a ball of fenestrated capillaries (i.e. capillaries which have larger-than-normal pores between the cells) surrounded by a capsular space. Substances can filter out through the pores in the capillaries; however, they have to be small enough. Hence, generally only unbound drug (i.e. drug not bound to a protein) can fit through- bound drugs tend to remain in the plasma. The glomerular filtration rate in an adult is 120mL/min, or 180L/day. The renal clearance by glomerular filtration can be calculated by multiplying this glomerular filtration rate by the fraction of the drug that is unbound in the plasma.
By the way, there's an acronym for the fraction of the unbound drug in plasma. It's fu. As our lecturer said, "Not meaning to be rude, but f u." (Oh, and for those of you who are also studying engineering or physics: my dad's a lecturer of electronic engineering, and he once told me that voltage across a diode is denoted by Vd. He finds himself often asking students to find the size of v D. You might have to read that one out loud to get the pun.)
The second process that occurs is active transport, which occurs in the proximal tubules of nephrons. Active transport, as I've mentioned before, involves the use of ATP-powered pumps to transport substances through a membrane. Actually, there are two membranes involved here, because the walls of the tubules are made of cells, and substances have to pass through the two opposite sides of the cell (the basolateral and apical sides) to get inside the nephron on the other side. As there are only a finite number of pumps, saturation can occur when all of the pumps are busy pumping stuff. Once saturation is reached, exaggerated pharmacological effects may be seen due to the extra drug remaining in the blood. This is what happens when people get drunk: their pumps get saturated.
There are two main types of pumps. One type includes the ABC Transporters, or ATP binding cassette. (I'm not sure what these do, to be honest.) The other type includes the SLC transporters, or solute carriers. These include OATs (organic anion transporters) and OCTs (organic cation transporters). These transporters can be blocked by certain drugs. Cimetidine blocks the cation transporters (they both start with c) and probenecid blocks OATs.
The third and final important process is diffusion, which occurs as substances are reabsorbed further down the tubules of the nephrons. Reabsorption is kinda important, because if the 120ml/min that was filtered through the glomerulus all reached our bladders, then we'd spend our lives tied to a toilet. Instead, only 1-2 ml/min of urine forms.
Reabsorption occurs because an increased concentration of substances in the nephron tubules may cause them to diffuse back into the blood down the concentration gradient. This occurs mainly for lipophilic and non-ionised drugs as they can cross the plasma membranes of the cells lining the tubules. The idea that non-ionised drugs reabsorb more easily is important in that this means that pH can affect the rate of reabsorption. You see, pH can affect whether ionisable groups are protonated or not, which in turn the charge on the molecule (see one of my earlier posts on this topic). If the pH is such that the molecule is charged, then it will have a very low rate of reabsorption.
Now to bring it all together! The "baseline" renal clearance is generally taken to be the clearance at the glomerulus, i.e. that old GFR (glomerular filtration rate) x fu (fraction unbound in plasma) that I alluded to earlier. However, if overall renal clearance is greater than this, this suggests that, overall, more drug must've entered the nephron past the glomerulus by means of active secretion. (Of course, some reabsorption may have occurred, but less was reabsorbed than secreted.) The opposite is true if overall renal clearance is smaller than GFR x fu: this indicates that more drug was reabsorbed than secreted.
4) Understand biliary excretion of drugs via the liver and the phenomenon of entero-hepatic recycling.
The liver is an important organ for excretion, partly because it's pretty much the first place a drug goes after being absorbed by the GI tract and partly because the liver's pretty awesome and has shitloads of functions, drug excretion being one of them. Liver cells have a lot of transporters and so forth which can move drugs and other substances into the bile duct. The bile duct leads to the duodenum (the very first part of the small intestines), and from there the drug eventually gets pooped out. Generally, drugs that are too large to be removed directly by the kidneys are eliminated via this mechanism.
It's not always as simple as the drug simply being crapped out, though. Some drugs undergo further metabolism in the GI tract. If these metabolites are lipophilic, they can be reabsorbed by the GI tract again and shipped back to the liver. This is a phenomenon known as enterohepatic recirculation. This lengthens the time that the drug stays in the body, and thus increases the time that the patient is exposed to the effects of the drug and/or the metabolite. This can be a problem if the metabolite is toxic.
5) Identify four tissues that play a minor role in drug excretion: skin, lungs, hair and breast milk.
The skin is a pretty minor route of excretion. Most of it happens through sweat, tears, saliva or desquamation (i.e. shedding of old skin cells). This is mainly an issue for miners and other people who have to do hard work with metals, as in these conditions more lead, cadmium, nickel and so forth may be sweated out than excreted via urine. High levels of sweat containing these metals can lead to dermatitis in sweaty areas such as the armpits and groin.
The lungs are somewhat important in secreting volatile substances, or compounds that have gaseous metabolites such as CO2. Secretion here occurs much the same as normal respiration- by gases in the blood diffusing into the alveoli and then exhaled.
Hair is a somewhat unintuitive one, as we don't often think of hair as a medium for excretion. However, many chemicals do deposit in the hair, and analysing hair segments can give an idea of the length of exposure to the chemical. This is useful for testing for illicit drug use.
Finally, breast milk is a minor route of excretion that can be concerning due to the risk of affecting the baby. Lipophilic and basic drugs can potentially pass into the fatty, acidic breast milk. In some cases, the nursing infant ends up getting a higher exposure to the drug (mg/kg) as compared to the mother.
So far, we've covered Absorption, Distribution and Metabolism- the first 3 letters of that ADME acronym that I've shown you before. Now I'm going to cover the final letter- Excretion!
1) Name and define the physiological processes that permanently rid the body of drugs and foreign chemicals.
Okay so I think this is just the definitions bit. There's a few important definitions that you have to know:
Excretion is the irreversible removal of the parent drug from the body. I *think* metabolism counts in this process, as the parent drug is being transformed into something that is... well, not the parent drug any more.
Elimination is the irreversible removal of the parent drug PLUS METABOLITES.
Clearance is the rate at which excretion occurs. (I think it's excretion rather than elimination anyway, as I'm fairly sure that the liver processing stuff is known as "hepatic clearance.") It is the volume of blood which is totally cleared of the drug per unit time and is thus measured in volume per time, such as litres per hour or millilitres per minute.
(Actually, I might need to double check some of these definitions. I'm currently asking a question on the PHAR2210 forums. I'll update this when I receive an answer- if I remember, anyway.)
One important acronym is fe: fraction excreted unchanged. It is the fraction of the drug that you pee out again in its original, unmetabolised form. (Sorry, I just put this here because I didn't know where else to put it.)
2) Identify organs and tissues playing the greatest role in removing drugs from the body.
The two main organs for removing drugs from the body are the liver and kidney. Other organs, such as the skin, lungs, hair and breasts (in lactating women) also play roles. I'll be covering all of these in greater detail throughout this post.
3) Identify three processes that control the excretion of drugs by the kidney.
The kidneys are a very important organ for removing drugs and their metabolites. Their functional units are called nephrons. Each nephron contains a glomerular capsule, which is where filtration takes place; proximal tubules, in which active secretion takes place; and the loop of Henle and distal tubules, where most reabsorption takes place.
The first process that occurs is filtration. The glomerular capsule, or Bowman's capsule, located at one "end" of the nephron, consists of a ball of fenestrated capillaries (i.e. capillaries which have larger-than-normal pores between the cells) surrounded by a capsular space. Substances can filter out through the pores in the capillaries; however, they have to be small enough. Hence, generally only unbound drug (i.e. drug not bound to a protein) can fit through- bound drugs tend to remain in the plasma. The glomerular filtration rate in an adult is 120mL/min, or 180L/day. The renal clearance by glomerular filtration can be calculated by multiplying this glomerular filtration rate by the fraction of the drug that is unbound in the plasma.
By the way, there's an acronym for the fraction of the unbound drug in plasma. It's fu. As our lecturer said, "Not meaning to be rude, but f u." (Oh, and for those of you who are also studying engineering or physics: my dad's a lecturer of electronic engineering, and he once told me that voltage across a diode is denoted by Vd. He finds himself often asking students to find the size of v D. You might have to read that one out loud to get the pun.)
The second process that occurs is active transport, which occurs in the proximal tubules of nephrons. Active transport, as I've mentioned before, involves the use of ATP-powered pumps to transport substances through a membrane. Actually, there are two membranes involved here, because the walls of the tubules are made of cells, and substances have to pass through the two opposite sides of the cell (the basolateral and apical sides) to get inside the nephron on the other side. As there are only a finite number of pumps, saturation can occur when all of the pumps are busy pumping stuff. Once saturation is reached, exaggerated pharmacological effects may be seen due to the extra drug remaining in the blood. This is what happens when people get drunk: their pumps get saturated.
There are two main types of pumps. One type includes the ABC Transporters, or ATP binding cassette. (I'm not sure what these do, to be honest.) The other type includes the SLC transporters, or solute carriers. These include OATs (organic anion transporters) and OCTs (organic cation transporters). These transporters can be blocked by certain drugs. Cimetidine blocks the cation transporters (they both start with c) and probenecid blocks OATs.
The third and final important process is diffusion, which occurs as substances are reabsorbed further down the tubules of the nephrons. Reabsorption is kinda important, because if the 120ml/min that was filtered through the glomerulus all reached our bladders, then we'd spend our lives tied to a toilet. Instead, only 1-2 ml/min of urine forms.
Reabsorption occurs because an increased concentration of substances in the nephron tubules may cause them to diffuse back into the blood down the concentration gradient. This occurs mainly for lipophilic and non-ionised drugs as they can cross the plasma membranes of the cells lining the tubules. The idea that non-ionised drugs reabsorb more easily is important in that this means that pH can affect the rate of reabsorption. You see, pH can affect whether ionisable groups are protonated or not, which in turn the charge on the molecule (see one of my earlier posts on this topic). If the pH is such that the molecule is charged, then it will have a very low rate of reabsorption.
Now to bring it all together! The "baseline" renal clearance is generally taken to be the clearance at the glomerulus, i.e. that old GFR (glomerular filtration rate) x fu (fraction unbound in plasma) that I alluded to earlier. However, if overall renal clearance is greater than this, this suggests that, overall, more drug must've entered the nephron past the glomerulus by means of active secretion. (Of course, some reabsorption may have occurred, but less was reabsorbed than secreted.) The opposite is true if overall renal clearance is smaller than GFR x fu: this indicates that more drug was reabsorbed than secreted.
4) Understand biliary excretion of drugs via the liver and the phenomenon of entero-hepatic recycling.
The liver is an important organ for excretion, partly because it's pretty much the first place a drug goes after being absorbed by the GI tract and partly because the liver's pretty awesome and has shitloads of functions, drug excretion being one of them. Liver cells have a lot of transporters and so forth which can move drugs and other substances into the bile duct. The bile duct leads to the duodenum (the very first part of the small intestines), and from there the drug eventually gets pooped out. Generally, drugs that are too large to be removed directly by the kidneys are eliminated via this mechanism.
It's not always as simple as the drug simply being crapped out, though. Some drugs undergo further metabolism in the GI tract. If these metabolites are lipophilic, they can be reabsorbed by the GI tract again and shipped back to the liver. This is a phenomenon known as enterohepatic recirculation. This lengthens the time that the drug stays in the body, and thus increases the time that the patient is exposed to the effects of the drug and/or the metabolite. This can be a problem if the metabolite is toxic.
5) Identify four tissues that play a minor role in drug excretion: skin, lungs, hair and breast milk.
The skin is a pretty minor route of excretion. Most of it happens through sweat, tears, saliva or desquamation (i.e. shedding of old skin cells). This is mainly an issue for miners and other people who have to do hard work with metals, as in these conditions more lead, cadmium, nickel and so forth may be sweated out than excreted via urine. High levels of sweat containing these metals can lead to dermatitis in sweaty areas such as the armpits and groin.
The lungs are somewhat important in secreting volatile substances, or compounds that have gaseous metabolites such as CO2. Secretion here occurs much the same as normal respiration- by gases in the blood diffusing into the alveoli and then exhaled.
Hair is a somewhat unintuitive one, as we don't often think of hair as a medium for excretion. However, many chemicals do deposit in the hair, and analysing hair segments can give an idea of the length of exposure to the chemical. This is useful for testing for illicit drug use.
Finally, breast milk is a minor route of excretion that can be concerning due to the risk of affecting the baby. Lipophilic and basic drugs can potentially pass into the fatty, acidic breast milk. In some cases, the nursing infant ends up getting a higher exposure to the drug (mg/kg) as compared to the mother.
Vertebral Column Evolution and Development
Now for another post on anatomy- this time on the development of the vertebral column! w00t w00t!
1. Evolution of the vertebral column
As you might be aware, humans belong to the phylum Chordata (i.e. the chordates). One of the defining features of chordates is the presence of a notochord during some stage of development.
For a bit more background, let's have a look at some more primitive types of chordates. Many simple chordates pretty much just sit on rocks and filter water to get their food. Chordate larvae can swim, however, and so they have a head, a notochord and a tail that extends past the anus. In these larvae, the notochord acts as a kind of "stiffening rod." When the larva moves, muscles on either side of the notochord contract alternately, allowing the tail to move. Eventually the larvae find rocks of their own to settle down on, and they lose the notochord and tail. They aren't "lost" completely though, as they're passed onto their next children. This phenomenon, in which characteristics only found in the juveniles are passed on, is known as "neotony."
Humans also belong to the subphylum Vertebrata, which as the name suggests, contains organisms that have a vertebral cord. I'm going to cover the development of the vertebral column throughout the rest of this post.
2. Stages in vertebral development
The first stage of vertebral development is the mesenchymatous stage, at around 4-6 weeks gestation. At this stage, neurulation and folding are complete, or pretty much complete. Body wall vessels also begin to appear between the somites.
During the mesenchymatous stage, sclerotome cells begin to migrate. Some surround and enclose the neural tube- these become the neural arch. Some move off to the sides- these become the costal elements. The third type, the perinotochordal cells, surround the notochord. The perinotochordal cells are also closer to the aforementioned body wall vessels and therefore get better nutrition and grow larger. The perinotochordal cells eventually form the vertebral bodies. (Don't worry, I'll explain what the neural arch, costal elements and bodies are later on.)
Since the perinotochordal cells surround the notochord, the notochord kind of gets "pinched off" into the space between somites. This is significant, as we'll see later.
The cartilaginous stage takes place at around 6-9 weeks gestation. During this stage, chondrification begins to occur as the mesenchyme is gradually replaced with hyaline cartilage. Paired centres of chondrification appear in the neural arch, costal elements and centrum (a.k.a. the body). Between vertebrae, fibrocartilage begins to form in a ring (the annulus fibrosus) around the notochordal cells (which will eventually become the nucleus pulposus of the intervertebral disc).
An interesting point to note here is that, since all the chondrification centres are paired, if issues arise with one member of a pair, asymmetrical vertebrae known as "hemivertebrae" can form.
The osseous stage occurs from around 8 weeks onwards. During this stage, the cartilage that formed in the previous stage begins to ossify (i.e. become bone). Once again, centres of ossification start to appear. The only difference this time is that there is only one centre of ossification in the centrum, rather than a pair (the neural arch and costal elements still have paired centres).
As the bone grows, cartilage growth plates continue to separate the three elements. An interlaminar growth plate separates the two neural arch elements (these eventually become the "laminae" of the vertebrae, hence the name "interlaminar"), while the neurocentral growth plate separates the neural arches and centrum (hence "neurocentral"). As for the costal elements, in the thorax they become ribs, whereas in the rest of the spinal column they fuse with the rest of the vertebrae to become part of the transverse processes (more on this in my next point).
Before I move on, though, I'll just make a note of things that can go wrong here. It's during the ossification stage that the location of notochordal cells can have an impact. If too many notochordal cells remain, then the centrum cannot ossify properly, resulting in "butterfly vertebrae" (so-called because apparently they look like butterflies). On the other hand, however, if there are too few notochordal cells between vertebrae, vertebrae can fuse together, forming "block vertebrae."
3. Regional differences in bone element contributions
Here is where I finally get to tell you what the neural arch, centrum and costal elements were all about!
So far, I've been using "centrum" and "body" pretty much interchangeably. That was kinda lazy of me, as the centrum doesn't actually end up forming the entire body of the vertebra. It forms most of it, but not all- the rest is made up of neural arch elements and, in the sacrum, parts of the costal elements as well.
As I've mentioned before, the costal elements form the ribs in thoracic vertebrae. In other vertebrae, they also help form parts of the transverse elements (the bits of the vertebrae that stick out to the sides).
Finally, the neural arch makes up pretty much everything else in the vertebrae, particularly the spinous processes (i.e. the bumpy bits at the back of the vertebrae).
4. Growth of the vertebral column
As I've mentioned before, there are cartilage growth plates around in the developing vertebrae: interlaminar and neurocentral. Growth can continue here until these plates close, which occurs at around 6-8 years of age. However, there is still some cartilage left: the ends of the processes still have cartilage, as does the top and bottom of the vertebral bodies (i.e. the endplates).
During puberty, secondary centres of ossification start to appear at the remaining cartilaginous spots, i.e. at the spinous process, transverse processes and ring apophysis (cartilage surrounding the endplates). The ring apophysis centre in particular is responsible for growth of the vertebral column. All of these growth plates close up by adulthood, so unfortunately we can't keep on getting taller forever :(
1. Evolution of the vertebral column
As you might be aware, humans belong to the phylum Chordata (i.e. the chordates). One of the defining features of chordates is the presence of a notochord during some stage of development.
For a bit more background, let's have a look at some more primitive types of chordates. Many simple chordates pretty much just sit on rocks and filter water to get their food. Chordate larvae can swim, however, and so they have a head, a notochord and a tail that extends past the anus. In these larvae, the notochord acts as a kind of "stiffening rod." When the larva moves, muscles on either side of the notochord contract alternately, allowing the tail to move. Eventually the larvae find rocks of their own to settle down on, and they lose the notochord and tail. They aren't "lost" completely though, as they're passed onto their next children. This phenomenon, in which characteristics only found in the juveniles are passed on, is known as "neotony."
Humans also belong to the subphylum Vertebrata, which as the name suggests, contains organisms that have a vertebral cord. I'm going to cover the development of the vertebral column throughout the rest of this post.
2. Stages in vertebral development
The first stage of vertebral development is the mesenchymatous stage, at around 4-6 weeks gestation. At this stage, neurulation and folding are complete, or pretty much complete. Body wall vessels also begin to appear between the somites.
During the mesenchymatous stage, sclerotome cells begin to migrate. Some surround and enclose the neural tube- these become the neural arch. Some move off to the sides- these become the costal elements. The third type, the perinotochordal cells, surround the notochord. The perinotochordal cells are also closer to the aforementioned body wall vessels and therefore get better nutrition and grow larger. The perinotochordal cells eventually form the vertebral bodies. (Don't worry, I'll explain what the neural arch, costal elements and bodies are later on.)
Since the perinotochordal cells surround the notochord, the notochord kind of gets "pinched off" into the space between somites. This is significant, as we'll see later.
The cartilaginous stage takes place at around 6-9 weeks gestation. During this stage, chondrification begins to occur as the mesenchyme is gradually replaced with hyaline cartilage. Paired centres of chondrification appear in the neural arch, costal elements and centrum (a.k.a. the body). Between vertebrae, fibrocartilage begins to form in a ring (the annulus fibrosus) around the notochordal cells (which will eventually become the nucleus pulposus of the intervertebral disc).
An interesting point to note here is that, since all the chondrification centres are paired, if issues arise with one member of a pair, asymmetrical vertebrae known as "hemivertebrae" can form.
The osseous stage occurs from around 8 weeks onwards. During this stage, the cartilage that formed in the previous stage begins to ossify (i.e. become bone). Once again, centres of ossification start to appear. The only difference this time is that there is only one centre of ossification in the centrum, rather than a pair (the neural arch and costal elements still have paired centres).
As the bone grows, cartilage growth plates continue to separate the three elements. An interlaminar growth plate separates the two neural arch elements (these eventually become the "laminae" of the vertebrae, hence the name "interlaminar"), while the neurocentral growth plate separates the neural arches and centrum (hence "neurocentral"). As for the costal elements, in the thorax they become ribs, whereas in the rest of the spinal column they fuse with the rest of the vertebrae to become part of the transverse processes (more on this in my next point).
Before I move on, though, I'll just make a note of things that can go wrong here. It's during the ossification stage that the location of notochordal cells can have an impact. If too many notochordal cells remain, then the centrum cannot ossify properly, resulting in "butterfly vertebrae" (so-called because apparently they look like butterflies). On the other hand, however, if there are too few notochordal cells between vertebrae, vertebrae can fuse together, forming "block vertebrae."
3. Regional differences in bone element contributions
Here is where I finally get to tell you what the neural arch, centrum and costal elements were all about!
So far, I've been using "centrum" and "body" pretty much interchangeably. That was kinda lazy of me, as the centrum doesn't actually end up forming the entire body of the vertebra. It forms most of it, but not all- the rest is made up of neural arch elements and, in the sacrum, parts of the costal elements as well.
As I've mentioned before, the costal elements form the ribs in thoracic vertebrae. In other vertebrae, they also help form parts of the transverse elements (the bits of the vertebrae that stick out to the sides).
Finally, the neural arch makes up pretty much everything else in the vertebrae, particularly the spinous processes (i.e. the bumpy bits at the back of the vertebrae).
4. Growth of the vertebral column
As I've mentioned before, there are cartilage growth plates around in the developing vertebrae: interlaminar and neurocentral. Growth can continue here until these plates close, which occurs at around 6-8 years of age. However, there is still some cartilage left: the ends of the processes still have cartilage, as does the top and bottom of the vertebral bodies (i.e. the endplates).
During puberty, secondary centres of ossification start to appear at the remaining cartilaginous spots, i.e. at the spinous process, transverse processes and ring apophysis (cartilage surrounding the endplates). The ring apophysis centre in particular is responsible for growth of the vertebral column. All of these growth plates close up by adulthood, so unfortunately we can't keep on getting taller forever :(
Sunday, March 27, 2016
Drug Metabolism
In my previous post, I mentioned the ADME acronym outlining the four major processes that happen to drugs: Absorption, Distribution, Metabolism and Excretion. In the previous post, I covered absorption and distribution. Now I'm going to cover metabolism- hence the title of this post.
1) Show an understanding of the term “biotransformation” and the importance of this phenomenon to mammalian organisms.
"Biotransformation," or metabolism, is the modification of drugs and other substances by chemicals in the body. This is important for several reasons. Firstly, just like enalapril, which I mentioned in my previous post, less active or inactive forms of a drug can be converted to more active forms. Secondly, biotransformation often serves to make drugs easier for the body to excrete, often by making them more water-soluble (for excretion in the kidneys). This prevents bioaccumulation and toxicity.
There are many types of reactions involved in biotransformation. Many of the enzymes required are located in the endoplasmic reticulum of liver cells, but they may be found in other locations as well. The most common types of reactions are oxidation, sulfation and glucuronidation (the addition of glucuronic acid, which is a carboxylic acid derived from glucose). Other less common reactions include acetylation, glutathione conjugation (glutathione is a tripeptide), methylation, amino acid conjugation, reduction and hydrolysis.
2) Identify the main classes of oxidative drug metabolism, showing an understanding of the CYP450 system in terms of basic enzymology, genetics and role in human drug metabolism.
There are two main types of oxidative drug metabolism: one in which oxygen is added and one in which hydrogen is removed. An example of an enzyme that catalyses the first type is Cytochrome P450, which I'm going to talk about very shortly. An example of an enzyme that catalyses the first type is alcohol dehydrogenase, which apparently we get to play with in the lab this week. Yay?
Okay, so back to the topic of Cytochrome P450, or CYP450 for short. There are four main families of CYP450, imaginatively named CYP1, CYP2, CYP3 and CYP4. The first three are the most important in the metabolism of xenobiotic (i.e. foreign substances like drugs) substances, while CYP4 is mostly used in the metabolism of fatty acids. All four types contain a Fe atom attached to a haem group, kinda like haemoglobin. There are also many different subtypes (a.k.a. isoforms) of CYP450. One of the most important CYP450 isoforms in human drug metabolism is called CYP3A4.
In order for CYP450 to do its job, it requires the electron carrier NADPH (which has made a cameo appearance in my posts before) and oxygen gas. CYP450 is located with NADPH oxidoreductase in the endoplasmic reticulum of liver cells. I have a very limited understanding of how this works but the NADPH oxidoreductase appears to move an electron from NADPH to CYP450, allowing it to carry out its job. In the end one of the oxygen atoms from O2 inserts into the drug, while the other joins with the H+ from NADPH to form water.
The activity of CYP450 can be induced by the introduction of drugs and certain other substances. Essentially, drugs bind to certain receptors in the cytosol known as xenosensors. This drug-xenosensor complex can then enter the nucleus, where it acts as a transcription factor and induces the synthesis and thus increases the activity of CYP450. It usually takes around a week for the effects to be seen. These effects include faster metabolism of the drug, which means that the drug leaves the body more rapidly. Hence more doses of the drug may need to be taken to have the desired effect.
CYP450, like other enzymes in the body, is also prone to competitive inhibition: when multiple substances are "competing" for the same enzyme. This may slow down or completely inhibit metabolism of drugs. In fact, CYP450 inhibition is a major cause of drug-drug interactions (DDIs). When metabolism is slowed down, the body may not be able to eliminate the drug as rapidly, leading to accumulation and toxicity. Alternatively, the body may not be able to convert an inactive drug into its active form, so that the patient may not experience the benefits.
An example of this CYP450 inhibition is the coadministration of fluoxetine (Prozac) and codeine. Fluoxetine inhibits CYP2D6, one of the isoforms of CYP450. Codeine, however, also relies on CYP2D6 to be converted into morphine and eventually morphine-6-glucuronide, both of which are actually responsible for the analgesic effect of codeine. Hence, coadministration of the two drugs means that codeine doesn't get converted into morphine and the patient doesn't experience relief from their pain.
CYP inhibition sounds annoying, but it can actually be used to have a beneficial effect. For example, Stribild, which is a cocktail of anti-HIV drugs, includes a CYP450 inhibitor. This CYP450 inhibitor stops the other drugs from being metabolised too quickly, so the patient only needs to take Stribild once a day.
There are several other factors that can affect the action of CYP450. One of these is genetic polymorphisms, such as deletions, insertions and repeats in the CYP450 gene. This leads to the rise of normal metabolisers (NM), poor metabolisers (PM), intermediate metabolisers (IM) and ultra metabolisers (UM) within a population.
3) Identify the main classes of conjugative drug metabolism, including sulfation & glucuronidation.
Conjugation is simply the addition of another molecule or substituent onto a drug. I've already mentioned several types, but I'm going to go into slightly more detail on sulfation and glucuronidation as they are the most common types of conjugative reactions. (By more detail I mean I'm just going to list some enzymes and cofactors. Enjoy.)
As previously mentioned, glucuronidation is the addition of glucuronic acid onto a drug. It is catalysed by UDP-glucuronosyltransferase, which uses UDP-glucuronic acid as a cofactor.
Sulfation is literally just the addition of a sulfate group (SO3). It usually happens on -OH and -NH2 groups. It is catalysed by sulfotransferase, which is located in the cytosol of certain cells. The cofactor here is PAPS (3'-phosphoadenosine 5'-phosphosulfate), which donates the sulfate group. (I've mentioned PAPS before in an earlier post about protein modifications.) Products formed from sulfation are usually very water-soluble.
1) Show an understanding of the term “biotransformation” and the importance of this phenomenon to mammalian organisms.
"Biotransformation," or metabolism, is the modification of drugs and other substances by chemicals in the body. This is important for several reasons. Firstly, just like enalapril, which I mentioned in my previous post, less active or inactive forms of a drug can be converted to more active forms. Secondly, biotransformation often serves to make drugs easier for the body to excrete, often by making them more water-soluble (for excretion in the kidneys). This prevents bioaccumulation and toxicity.
There are many types of reactions involved in biotransformation. Many of the enzymes required are located in the endoplasmic reticulum of liver cells, but they may be found in other locations as well. The most common types of reactions are oxidation, sulfation and glucuronidation (the addition of glucuronic acid, which is a carboxylic acid derived from glucose). Other less common reactions include acetylation, glutathione conjugation (glutathione is a tripeptide), methylation, amino acid conjugation, reduction and hydrolysis.
2) Identify the main classes of oxidative drug metabolism, showing an understanding of the CYP450 system in terms of basic enzymology, genetics and role in human drug metabolism.
There are two main types of oxidative drug metabolism: one in which oxygen is added and one in which hydrogen is removed. An example of an enzyme that catalyses the first type is Cytochrome P450, which I'm going to talk about very shortly. An example of an enzyme that catalyses the first type is alcohol dehydrogenase, which apparently we get to play with in the lab this week. Yay?
Okay, so back to the topic of Cytochrome P450, or CYP450 for short. There are four main families of CYP450, imaginatively named CYP1, CYP2, CYP3 and CYP4. The first three are the most important in the metabolism of xenobiotic (i.e. foreign substances like drugs) substances, while CYP4 is mostly used in the metabolism of fatty acids. All four types contain a Fe atom attached to a haem group, kinda like haemoglobin. There are also many different subtypes (a.k.a. isoforms) of CYP450. One of the most important CYP450 isoforms in human drug metabolism is called CYP3A4.
In order for CYP450 to do its job, it requires the electron carrier NADPH (which has made a cameo appearance in my posts before) and oxygen gas. CYP450 is located with NADPH oxidoreductase in the endoplasmic reticulum of liver cells. I have a very limited understanding of how this works but the NADPH oxidoreductase appears to move an electron from NADPH to CYP450, allowing it to carry out its job. In the end one of the oxygen atoms from O2 inserts into the drug, while the other joins with the H+ from NADPH to form water.
The activity of CYP450 can be induced by the introduction of drugs and certain other substances. Essentially, drugs bind to certain receptors in the cytosol known as xenosensors. This drug-xenosensor complex can then enter the nucleus, where it acts as a transcription factor and induces the synthesis and thus increases the activity of CYP450. It usually takes around a week for the effects to be seen. These effects include faster metabolism of the drug, which means that the drug leaves the body more rapidly. Hence more doses of the drug may need to be taken to have the desired effect.
CYP450, like other enzymes in the body, is also prone to competitive inhibition: when multiple substances are "competing" for the same enzyme. This may slow down or completely inhibit metabolism of drugs. In fact, CYP450 inhibition is a major cause of drug-drug interactions (DDIs). When metabolism is slowed down, the body may not be able to eliminate the drug as rapidly, leading to accumulation and toxicity. Alternatively, the body may not be able to convert an inactive drug into its active form, so that the patient may not experience the benefits.
An example of this CYP450 inhibition is the coadministration of fluoxetine (Prozac) and codeine. Fluoxetine inhibits CYP2D6, one of the isoforms of CYP450. Codeine, however, also relies on CYP2D6 to be converted into morphine and eventually morphine-6-glucuronide, both of which are actually responsible for the analgesic effect of codeine. Hence, coadministration of the two drugs means that codeine doesn't get converted into morphine and the patient doesn't experience relief from their pain.
CYP inhibition sounds annoying, but it can actually be used to have a beneficial effect. For example, Stribild, which is a cocktail of anti-HIV drugs, includes a CYP450 inhibitor. This CYP450 inhibitor stops the other drugs from being metabolised too quickly, so the patient only needs to take Stribild once a day.
There are several other factors that can affect the action of CYP450. One of these is genetic polymorphisms, such as deletions, insertions and repeats in the CYP450 gene. This leads to the rise of normal metabolisers (NM), poor metabolisers (PM), intermediate metabolisers (IM) and ultra metabolisers (UM) within a population.
3) Identify the main classes of conjugative drug metabolism, including sulfation & glucuronidation.
Conjugation is simply the addition of another molecule or substituent onto a drug. I've already mentioned several types, but I'm going to go into slightly more detail on sulfation and glucuronidation as they are the most common types of conjugative reactions. (By more detail I mean I'm just going to list some enzymes and cofactors. Enjoy.)
As previously mentioned, glucuronidation is the addition of glucuronic acid onto a drug. It is catalysed by UDP-glucuronosyltransferase, which uses UDP-glucuronic acid as a cofactor.
Sulfation is literally just the addition of a sulfate group (SO3). It usually happens on -OH and -NH2 groups. It is catalysed by sulfotransferase, which is located in the cytosol of certain cells. The cofactor here is PAPS (3'-phosphoadenosine 5'-phosphosulfate), which donates the sulfate group. (I've mentioned PAPS before in an earlier post about protein modifications.) Products formed from sulfation are usually very water-soluble.
Saturday, March 26, 2016
Drug Absorption and Distribution
Yup, I'm doing Pharmacology this semester :) This course starts off with ADME- Absorption, Distribution, Metabolism and Excretion. It's all to do with what happens to a drug once you put it into the body.
1) Explain what the term “pharmacokinetics” means and identify the 4 phases that control the fate of drugs in the body
"Pharmacokinetics" is essentially "what the body does to the drug." When you take a drug, it's not all about the effects that the drug has on the body (i.e. its pharmacodynamics)- the body may have to do stuff to it to get it to work, and the body will also have to do stuff to get rid of the drug later. The four phases are those mentioned previously: absorption, distribution, metabolism and excretion, all summed up with the acronym ADME.
2) Understand the process of drug absorption, the organs in which it occurs, the factors that govern drug uptake at absorption sites.
Absorption is the uptake of a drug into the systemic circulation. Uptake of many drugs occurs in the GI tract, since so many drugs are taken orally. Drugs are taken up into the cells through pretty much the same mechanisms in which nearly every substance is taken up into the cells: through diffusion, facilitated diffusion, active transport, phagocytosis etc. The factors that govern uptake are pretty similar to the factors that govern uptake of any other substance through these methods: highly polarised molecules don't diffuse well through the lipid membrane, facilitated diffusion and active transport require the presence of specialised proteins etc.
I'm now going to focus on three main factors that govern drug intake: size, solubility and external pH. I'll start with size: the larger the drug, the less well absorbed it is. Some dude called Lipinski once said that for optimal oral absorption, the molecular weight of a drug should be no larger than 500 g/mol.
Solubility is a bit more complicated. A highly hydrophilic drug will travel through the blood fairly readily, but it won't pass through lipid membranes as well. The opposite is true for lipophilic drugs. Most of the time, a happy medium needs to be found between these two extremes. There are two main measures used to estimate solubility in aqueous and lipid solvents: LogP and polar surface area.
LogP is the log of the partition coefficient of the drug. The partition coefficient is the ratio between the percentage of a drug that dissolves in water versus the percentage that dissolves in a lipid solvent such as octanol. LogP predicts the ability of a drug to cross a lipid membrane.
Polar surface area is simply a way at comparing the size of polar and non-polar areas on the surface of a molecule. (If you don't know much about polarity, check out my post on intermolecular bonding.) Usually this is done by a computer.
Some guy called Egan made a plot of the LogP and polar surface areas of many drugs on the market. He found that all drugs lie within a roughly oval-shaped plot on this graph. This is known as "Egan's Egg."
The reasons why external pH affects the absorption of a drug is pretty much related to the solubility. You see, many drugs have ionisable groups that may carry a charge depending on the pH (I've mentioned this before in a different context- see my earlier post on acid-base chemistry of amino acids.) If the drug is charged, then it is less likely to cross the lipid membranes of cells.
3) Understand the concept of a “prodrug.”
Absorption of drugs can be changed by adding different substituents to them. Different substituents can increase or decrease hydrophobicity (which is related to the ability to cross the membrane). The amount that the hydrophobicity increases or decreases is known as the hydrophobicity constant of that substituent.
These substituents can not only alter hydrophobicity on their own, but they can also have an effect by attaching directly to hydrophobic or hydrophilic groups originally on the drug, "masking" their effects. These "masked drugs" are also known as "prodrugs." When the drug enters the target cell, enzymes within the target cell can cleave off the groups to release the active drug.
An example of a "prodrug" is enalapril, which is a blood pressure medication. Enalapril is an inactive prodrug that contains an ester linkage at one point. Once in the body, several esterases can cleave this ester linkage to "unmask" an -OH group, forming enalaprilat, which actually does the job. Similar mechanisms can potentially also be used to ensure that drugs only have an effect in the target organ, if you can find "masking groups" that are only removed by enzymes in the target organ.
4) Explain what is meant by the term “bioavailability” and the factors that contribute to this process.
The bioavailability of a drug is the amount that reaches the systemic circulation. Of course, this is affected by the absorption processes outlined above. Bioavailability is also affected by the hepatic clearance of the drug (as stuff from the GI tract is taken to the liver for processing), though this is a topic for a later post.
A quick, somewhat important point to make is that the presence of active transporters in the GI tract may affect bioavailability. For example, p-glycoprotein is a transporter that transports many drugs back into the lumen of the GI tract. This is useful for protecting us against toxins, but not so useful when we actually want the drug to be absorbed and have an effect.
5) Show an awareness of the distribution of drugs within the body, and the tendency for particular drugs to accumulate in specific tissues.
As alluded to before, some drugs are more hydrophobic or hydrophilic than others. This can determine where drugs tend to "hang out." Hydrophilic drugs such as warfarin tend to stay in the blood and extracellular fluid and thus yield high plasma concentrations, whereas lipophilic drugs tend to yield much lower plasma concentrations as they are more readily absorbed by the tissues. This gives rise to a theoretical concept known as "Volume of Distribution" which can help to determine whether a drug is more or less readily absorbed by the tissues. Volume of Distribution can be calculated by dividing the dose in milligrams by the plasma concentration of the drug in mg/L:
Vdist (L) = dose (mg) / plasma concentration (mg/L)
Note that the volume of distribution is not an actual value, just an imaginary one to help us figure out where the drug goes. Some drugs have a volume of distribution much larger than the total volume of fluid in the body.
To explain how this concept "works," let's take a hypothetical drug that has a dose of 100mg and a plasma concentration of 10mg/L. That plasma concentration of 10mg/L is just like dissolving that original 100mg into 10L of water. Hence the volume of distribution is 10L.
Very hydrophilic drugs tend to have a low volume of distribution and are found mainly in the plasma, but as you increase the lipophilicity of a drug, the drug will find its way into interstitial and then into intracellular fluids, and so on.
Finally, a quick note on protein binding and how this affects where drugs remain. The blood contains several proteins such as albumin and alpha-glycoprotein, which help to carry drugs around the blood. Albumin mainly carries acidic drugs, whereas alpha-glycoprotein tends to carry basic drugs. This binding tends to confine the drug to plasma. It also makes extraction and excretion by the kidneys and liver a bit harder.
Tissues also have binding proteins of their own, and these can be especially significant in skeletal muscle for some drugs. The equilibrium between the free and bound drug in the tissues and plasma can complicate the distribution of drugs.
Some drugs, particularly very lipophilic ones, may also be stored in fat. This can cause issues in cases of obesity.
1) Explain what the term “pharmacokinetics” means and identify the 4 phases that control the fate of drugs in the body
"Pharmacokinetics" is essentially "what the body does to the drug." When you take a drug, it's not all about the effects that the drug has on the body (i.e. its pharmacodynamics)- the body may have to do stuff to it to get it to work, and the body will also have to do stuff to get rid of the drug later. The four phases are those mentioned previously: absorption, distribution, metabolism and excretion, all summed up with the acronym ADME.
2) Understand the process of drug absorption, the organs in which it occurs, the factors that govern drug uptake at absorption sites.
Absorption is the uptake of a drug into the systemic circulation. Uptake of many drugs occurs in the GI tract, since so many drugs are taken orally. Drugs are taken up into the cells through pretty much the same mechanisms in which nearly every substance is taken up into the cells: through diffusion, facilitated diffusion, active transport, phagocytosis etc. The factors that govern uptake are pretty similar to the factors that govern uptake of any other substance through these methods: highly polarised molecules don't diffuse well through the lipid membrane, facilitated diffusion and active transport require the presence of specialised proteins etc.
I'm now going to focus on three main factors that govern drug intake: size, solubility and external pH. I'll start with size: the larger the drug, the less well absorbed it is. Some dude called Lipinski once said that for optimal oral absorption, the molecular weight of a drug should be no larger than 500 g/mol.
Solubility is a bit more complicated. A highly hydrophilic drug will travel through the blood fairly readily, but it won't pass through lipid membranes as well. The opposite is true for lipophilic drugs. Most of the time, a happy medium needs to be found between these two extremes. There are two main measures used to estimate solubility in aqueous and lipid solvents: LogP and polar surface area.
LogP is the log of the partition coefficient of the drug. The partition coefficient is the ratio between the percentage of a drug that dissolves in water versus the percentage that dissolves in a lipid solvent such as octanol. LogP predicts the ability of a drug to cross a lipid membrane.
Polar surface area is simply a way at comparing the size of polar and non-polar areas on the surface of a molecule. (If you don't know much about polarity, check out my post on intermolecular bonding.) Usually this is done by a computer.
Some guy called Egan made a plot of the LogP and polar surface areas of many drugs on the market. He found that all drugs lie within a roughly oval-shaped plot on this graph. This is known as "Egan's Egg."
The reasons why external pH affects the absorption of a drug is pretty much related to the solubility. You see, many drugs have ionisable groups that may carry a charge depending on the pH (I've mentioned this before in a different context- see my earlier post on acid-base chemistry of amino acids.) If the drug is charged, then it is less likely to cross the lipid membranes of cells.
3) Understand the concept of a “prodrug.”
Absorption of drugs can be changed by adding different substituents to them. Different substituents can increase or decrease hydrophobicity (which is related to the ability to cross the membrane). The amount that the hydrophobicity increases or decreases is known as the hydrophobicity constant of that substituent.
These substituents can not only alter hydrophobicity on their own, but they can also have an effect by attaching directly to hydrophobic or hydrophilic groups originally on the drug, "masking" their effects. These "masked drugs" are also known as "prodrugs." When the drug enters the target cell, enzymes within the target cell can cleave off the groups to release the active drug.
An example of a "prodrug" is enalapril, which is a blood pressure medication. Enalapril is an inactive prodrug that contains an ester linkage at one point. Once in the body, several esterases can cleave this ester linkage to "unmask" an -OH group, forming enalaprilat, which actually does the job. Similar mechanisms can potentially also be used to ensure that drugs only have an effect in the target organ, if you can find "masking groups" that are only removed by enzymes in the target organ.
4) Explain what is meant by the term “bioavailability” and the factors that contribute to this process.
The bioavailability of a drug is the amount that reaches the systemic circulation. Of course, this is affected by the absorption processes outlined above. Bioavailability is also affected by the hepatic clearance of the drug (as stuff from the GI tract is taken to the liver for processing), though this is a topic for a later post.
A quick, somewhat important point to make is that the presence of active transporters in the GI tract may affect bioavailability. For example, p-glycoprotein is a transporter that transports many drugs back into the lumen of the GI tract. This is useful for protecting us against toxins, but not so useful when we actually want the drug to be absorbed and have an effect.
5) Show an awareness of the distribution of drugs within the body, and the tendency for particular drugs to accumulate in specific tissues.
As alluded to before, some drugs are more hydrophobic or hydrophilic than others. This can determine where drugs tend to "hang out." Hydrophilic drugs such as warfarin tend to stay in the blood and extracellular fluid and thus yield high plasma concentrations, whereas lipophilic drugs tend to yield much lower plasma concentrations as they are more readily absorbed by the tissues. This gives rise to a theoretical concept known as "Volume of Distribution" which can help to determine whether a drug is more or less readily absorbed by the tissues. Volume of Distribution can be calculated by dividing the dose in milligrams by the plasma concentration of the drug in mg/L:
Vdist (L) = dose (mg) / plasma concentration (mg/L)
Note that the volume of distribution is not an actual value, just an imaginary one to help us figure out where the drug goes. Some drugs have a volume of distribution much larger than the total volume of fluid in the body.
To explain how this concept "works," let's take a hypothetical drug that has a dose of 100mg and a plasma concentration of 10mg/L. That plasma concentration of 10mg/L is just like dissolving that original 100mg into 10L of water. Hence the volume of distribution is 10L.
Very hydrophilic drugs tend to have a low volume of distribution and are found mainly in the plasma, but as you increase the lipophilicity of a drug, the drug will find its way into interstitial and then into intracellular fluids, and so on.
Finally, a quick note on protein binding and how this affects where drugs remain. The blood contains several proteins such as albumin and alpha-glycoprotein, which help to carry drugs around the blood. Albumin mainly carries acidic drugs, whereas alpha-glycoprotein tends to carry basic drugs. This binding tends to confine the drug to plasma. It also makes extraction and excretion by the kidneys and liver a bit harder.
Tissues also have binding proteins of their own, and these can be especially significant in skeletal muscle for some drugs. The equilibrium between the free and bound drug in the tissues and plasma can complicate the distribution of drugs.
Some drugs, particularly very lipophilic ones, may also be stored in fat. This can cause issues in cases of obesity.
Friday, March 25, 2016
Embryology and Evolution
I wasn't going to do a third post about this in a single day, but I shouldn't have too much more to add on this topic *fingers crossed.* Besides, I just looked at the sample exam questions and damn near had a panic attack. So here goes...
So basically the main theme of this post is "ontogeny recapitulates phylogeny." Ontogeny is the development of the individual, and phylogeny is the development of the species. The idea is that as the embryo develops, it moves from a primitive one-cell organism and then gets more and more complex until it becomes a human or other animal, and this mirrors the development of organisms from simple one-celled organisms to more and more complex things. The idea isn't perfect, but it's a useful concept in understanding how many structures develop. In this post, we're going to see how this relates to the pharyngeal arches of the embryo.
In primitive jawless fish, the pharyngeal arches are the gill arches, with the first arch making up the mouth. In less-primitive fish with jaws, the first arch makes up the upper and lower jaws. The lower jaw is made up of several parts: the dentary (holds the teeth) and the articular and quadrate (form the jaw joint).
In mammals, like us, our first pharyngeal arch also develops into the mouth. Instead of the dentary, we have Meckel's cartilage, which also serves to hold the teeth. It develops into the mandible, and eventually articulates with the temporal bone but only after the articular and quadrate cartilages have buggered off. Where do they bugger off to? Well, the articular cartilage forms the malleus ("hammer") bone of the middle ear while the quadrate cartilage forms the incus of the middle ear. The other arches contribute to other structures of the neck, such as the larynx, but I won't go into those now.
And, um, that's all I need to cover on this topic. Main point is that the development of our jaw starts off very much like the development of the jaw in fish and reptiles. But then other stuff happens because we're more complex. Or something.
So basically the main theme of this post is "ontogeny recapitulates phylogeny." Ontogeny is the development of the individual, and phylogeny is the development of the species. The idea is that as the embryo develops, it moves from a primitive one-cell organism and then gets more and more complex until it becomes a human or other animal, and this mirrors the development of organisms from simple one-celled organisms to more and more complex things. The idea isn't perfect, but it's a useful concept in understanding how many structures develop. In this post, we're going to see how this relates to the pharyngeal arches of the embryo.
In primitive jawless fish, the pharyngeal arches are the gill arches, with the first arch making up the mouth. In less-primitive fish with jaws, the first arch makes up the upper and lower jaws. The lower jaw is made up of several parts: the dentary (holds the teeth) and the articular and quadrate (form the jaw joint).
In mammals, like us, our first pharyngeal arch also develops into the mouth. Instead of the dentary, we have Meckel's cartilage, which also serves to hold the teeth. It develops into the mandible, and eventually articulates with the temporal bone but only after the articular and quadrate cartilages have buggered off. Where do they bugger off to? Well, the articular cartilage forms the malleus ("hammer") bone of the middle ear while the quadrate cartilage forms the incus of the middle ear. The other arches contribute to other structures of the neck, such as the larynx, but I won't go into those now.
And, um, that's all I need to cover on this topic. Main point is that the development of our jaw starts off very much like the development of the jaw in fish and reptiles. But then other stuff happens because we're more complex. Or something.
Embryology 2: Fate of the Ectoderm and Mesoderm
After covering a lot in my first post on embryology, I shouldn't have to type out too much for this next post. At least, I hope I don't have to type out too much, anyway...
Ectoderm – Surface Ectoderm – Neural tube – Neural crest
In my previous post, I wrote about neurulation (i.e. the development of the neural tube and neural crest) and in the list at the end I wrote a bit about what those structures can develop into. There's not a lot more to develop on that, which means less reading for you and less typing for me :D
The anterior pituitary is one of the structures that develops from the surface ectoderm, as previously listed. It does this by folding inwards to create a structure known as Rathke's pouch. Rathke's pouch then detaches and moves next to the infundibulum of the developing brain (it's essentially a protrusion on the base of the brain, to my understanding) to form the anterior part of the pituitary gland.
I'm just going to change topics a bit here to talk about the oral and cloacal membranes. One thing I forgot to mention in my last post is that the mesoderm doesn't completely separate the endoderm and ectoderm. Instead, there are two places where the endoderm and ectoderm remain directly in contact. One place is near the cranial end, and is known as the oral membrane. The other place is near the caudal end, and is known as the cloacal membrane. The oral membrane eventually ruptures through and forms the oral cavity, whereas the cloacal membrane forms the anal, genital and urinary openings when it ruptures through.
The reason I bring this up now is because the development of Rathke's pouch and thus the anterior pituitary also highlights the fact that the oral membrane is behind the mouth. Confused? Well, since the oral membrane is the location where the ectoderm and endoderm meet, and it ruptures through to form the oral cavity, anything past the oral membrane is formed from endoderm (like the lining of your oesophagus and digestive system). However, the anterior pituitary, which is formed from cells lining the mouth, is ectodermal in origin. Hence the mouth, and the lining of the mouth, must be in front of the oral membrane.
Now I've spoken about surface ectoderm, I'm going to move on to neural tube ectoderm. As mentioned in my previous post, it's responsible for the formation of the central nervous system- that is, the brain and spinal cord. The neural tube is expanded at the cranial end, which forms the brain. The canal that runs through the neural tube eventually becomes the ventricles of the brain (cerebrospinal fluid flows through the ventricles). Motor neurons are located on the ventral side of the canal whereas sensory neurons are located on the dorsal side.
The canal is made up of grey matter and white matter, named due to their appearance. Grey matter is comprised of nerve cell bodies, while white matter is comprised of myelinated axon sheaths (myelin looks white, hence the name). In the part of the canal that eventually becomes the spinal cord, grey matter is found more on the inside lining the canal, while white matter is found on the outside. This situation is reversed in the brain.
Finally, a quick word on the neural crest ectoderm. There's not much I can expand on here, other than giving some more examples of things that the neural crest ectoderm eventually becomes. Aside from the sensory epithelia of the eyes, ears and nose, peripheral nervous system and adrenal medulla (yup, I copy-pasted that list from the previous post and edited in the word "and"), the neural crest cells also develop into Schwann cells (oligodendrocytes which form the myelin sheath in the peripheral nervous system), the pia and arachnoid meninges (which are collectively known as "leptomeninges"), the dentine of the teeth, the cornea of the eyes and melanocytes (the cells that produce melanin). The neural crest also forms much of the cartilage, muscle and so forth in the head that were previously thought to have developed from mesoderm. And speaking of the head...
Development of the Head
Many structures in the head are simply continuations of structures in the rest of the body. For example, as previously mentioned, the neural tube continues up into the head to become the brain. The vertebral column also continues up to form the base of the skull. Many structures, such as the cranial vault, pharyngeal arches (precursors for structures such as the jaw, larynx and other structures in the neck) and sensory placodes (i.e. the eyes, ears and nose) develop via interaction between the neural tube or neural crest with the surface ectoderm.
Mesoderm – Paraxial – Intermediate – Lateral plate
I don't think I have much to say on the mesoderm. That being said, I didn't think that I had much to say on the ectoderm, either, and I wrote six paragraphs on it, not counting my whiny introductory paragraph.
I'll start with the lateral plate mesoderm, because I'm fairly sure my first post pretty much covered this. I did promise that I'd talk about the somatopleure and visceropleure a bit more, though, which was kind of stupid of me seeing that I don't really have much more to say about them. Both are responsible for the formation of smooth muscle, connective tissue and vasculature, but the somatopleure is responsible for the stuff associated with the epidermis of the body wall and limbs and the visceropleure is responsible for the stuff associated with the epithelia of the gut.
Now I'll talk a bit about the paraxial mesoderm, and this time I'm fairly sure that I do have something to say about it that I haven't said before: namely, the idea of the epimere and hypomere. Some cells of the dermatome and myotome remain put and are known as the epimere, while others migrate into the somatopleure and are known as the hypomere. (I'm not sure about sclerotome, though- maybe this is why last year they told us that sclerotome only forms the axial skeleton?) Cells of the epimere become the dermis and muscles of the back, while cells of the hypomere become the dermis and muscles of the body wall and limbs.
Before I get onto how this relates to nerve supply to different organs (because yes, this epimere/hypomere stuff relates to nerve supply to different organs), I'm first going to have a quick talk about typical spinal nerves. Behold, this picture that I so rudely took from http://thegoofyanatomist.weebly.com/uploads/3/0/9/9/30995885/1422050582.png:
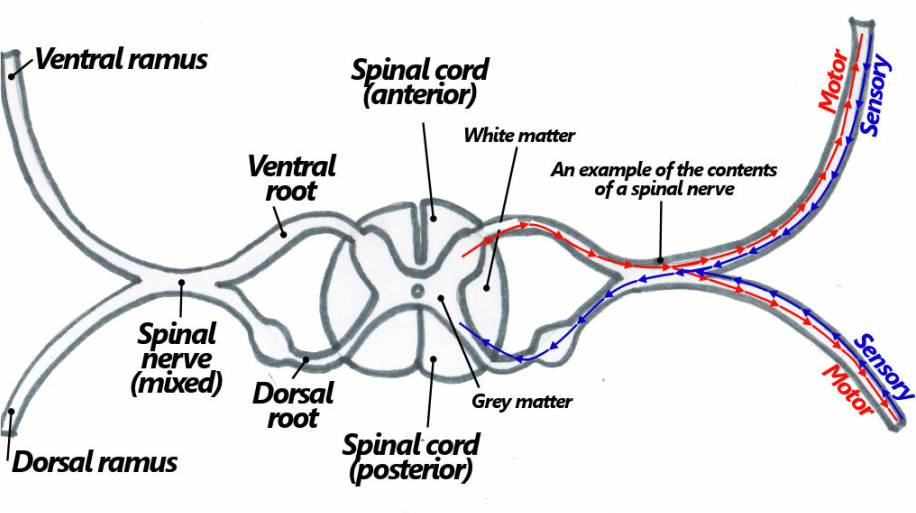

Limb Development
We didn't really cover limb development in that much detail (in fact I'm fairly sure the lecturer pretty much skipped over the slide relating to limb development as we were going overtime), so all I can really tell you are the basics. Limbs develop from the somatopleure, which as I've mentioned several times, is the layer of the lateral plate mesoderm that associates with the body wall. The muscle and skin in this area is from the hypomere and is therefore supplied by ventral rami. Limb development starts from little limb buds that begin to form in the 4th week, which induce the proliferation of underlying mesoderm to create the limb. Formation of limbs are proximodistal (i.e. from the "close" end of the limb to the "far" end).
Ectoderm – Surface Ectoderm – Neural tube – Neural crest
In my previous post, I wrote about neurulation (i.e. the development of the neural tube and neural crest) and in the list at the end I wrote a bit about what those structures can develop into. There's not a lot more to develop on that, which means less reading for you and less typing for me :D
The anterior pituitary is one of the structures that develops from the surface ectoderm, as previously listed. It does this by folding inwards to create a structure known as Rathke's pouch. Rathke's pouch then detaches and moves next to the infundibulum of the developing brain (it's essentially a protrusion on the base of the brain, to my understanding) to form the anterior part of the pituitary gland.
I'm just going to change topics a bit here to talk about the oral and cloacal membranes. One thing I forgot to mention in my last post is that the mesoderm doesn't completely separate the endoderm and ectoderm. Instead, there are two places where the endoderm and ectoderm remain directly in contact. One place is near the cranial end, and is known as the oral membrane. The other place is near the caudal end, and is known as the cloacal membrane. The oral membrane eventually ruptures through and forms the oral cavity, whereas the cloacal membrane forms the anal, genital and urinary openings when it ruptures through.
The reason I bring this up now is because the development of Rathke's pouch and thus the anterior pituitary also highlights the fact that the oral membrane is behind the mouth. Confused? Well, since the oral membrane is the location where the ectoderm and endoderm meet, and it ruptures through to form the oral cavity, anything past the oral membrane is formed from endoderm (like the lining of your oesophagus and digestive system). However, the anterior pituitary, which is formed from cells lining the mouth, is ectodermal in origin. Hence the mouth, and the lining of the mouth, must be in front of the oral membrane.
Now I've spoken about surface ectoderm, I'm going to move on to neural tube ectoderm. As mentioned in my previous post, it's responsible for the formation of the central nervous system- that is, the brain and spinal cord. The neural tube is expanded at the cranial end, which forms the brain. The canal that runs through the neural tube eventually becomes the ventricles of the brain (cerebrospinal fluid flows through the ventricles). Motor neurons are located on the ventral side of the canal whereas sensory neurons are located on the dorsal side.
The canal is made up of grey matter and white matter, named due to their appearance. Grey matter is comprised of nerve cell bodies, while white matter is comprised of myelinated axon sheaths (myelin looks white, hence the name). In the part of the canal that eventually becomes the spinal cord, grey matter is found more on the inside lining the canal, while white matter is found on the outside. This situation is reversed in the brain.
Finally, a quick word on the neural crest ectoderm. There's not much I can expand on here, other than giving some more examples of things that the neural crest ectoderm eventually becomes. Aside from the sensory epithelia of the eyes, ears and nose, peripheral nervous system and adrenal medulla (yup, I copy-pasted that list from the previous post and edited in the word "and"), the neural crest cells also develop into Schwann cells (oligodendrocytes which form the myelin sheath in the peripheral nervous system), the pia and arachnoid meninges (which are collectively known as "leptomeninges"), the dentine of the teeth, the cornea of the eyes and melanocytes (the cells that produce melanin). The neural crest also forms much of the cartilage, muscle and so forth in the head that were previously thought to have developed from mesoderm. And speaking of the head...
Development of the Head
Many structures in the head are simply continuations of structures in the rest of the body. For example, as previously mentioned, the neural tube continues up into the head to become the brain. The vertebral column also continues up to form the base of the skull. Many structures, such as the cranial vault, pharyngeal arches (precursors for structures such as the jaw, larynx and other structures in the neck) and sensory placodes (i.e. the eyes, ears and nose) develop via interaction between the neural tube or neural crest with the surface ectoderm.
Mesoderm – Paraxial – Intermediate – Lateral plate
I don't think I have much to say on the mesoderm. That being said, I didn't think that I had much to say on the ectoderm, either, and I wrote six paragraphs on it, not counting my whiny introductory paragraph.
I'll start with the lateral plate mesoderm, because I'm fairly sure my first post pretty much covered this. I did promise that I'd talk about the somatopleure and visceropleure a bit more, though, which was kind of stupid of me seeing that I don't really have much more to say about them. Both are responsible for the formation of smooth muscle, connective tissue and vasculature, but the somatopleure is responsible for the stuff associated with the epidermis of the body wall and limbs and the visceropleure is responsible for the stuff associated with the epithelia of the gut.
Now I'll talk a bit about the paraxial mesoderm, and this time I'm fairly sure that I do have something to say about it that I haven't said before: namely, the idea of the epimere and hypomere. Some cells of the dermatome and myotome remain put and are known as the epimere, while others migrate into the somatopleure and are known as the hypomere. (I'm not sure about sclerotome, though- maybe this is why last year they told us that sclerotome only forms the axial skeleton?) Cells of the epimere become the dermis and muscles of the back, while cells of the hypomere become the dermis and muscles of the body wall and limbs.
Before I get onto how this relates to nerve supply to different organs (because yes, this epimere/hypomere stuff relates to nerve supply to different organs), I'm first going to have a quick talk about typical spinal nerves. Behold, this picture that I so rudely took from http://thegoofyanatomist.weebly.com/uploads/3/0/9/9/30995885/1422050582.png:
As I've mentioned before, sensory nerves are found on the dorsal (posterior) side of the cord, whereas motor nerves are found on the ventral (anterior) side of the cord. (Just in case you want to know, that bulge on the dorsal side is known as the "dorsal root ganglion," and it contains a cluster of sensory nerve cell bodies.) Sensory and motor neurons eventually meet to form a mixed spinal nerve, as you can see on the right side of the diagram. Mixed spinal nerves branch out into two divisions: the ventral rami and the dorsal rami. I'll get to them in a bit. Some nerves, specifically the nerves between T1 and L1, also have another "white ramus" which leads to a sympathetic ganglion of the sympathetic trunk. From here sympathetic nerves run to the body wall via the grey ramus, or to the viscera via visceral branches.
Now I'm going to get back to those ventral and dorsal rami. While the cells of the hypomere migrate, they take their own nerve supply with them. This nerve supply is the ventral rami. Hence, the epimere is supplied by the dorsal rami, while the hypomere is supplied by the ventral rami.
For some reason, when we were learning about this during the lecture, I had a mental image of muscle cells (representing myotome) marching to the ventral side of the embryo dragging nerves behind them. No, seriously, here's the picture that I doodled during the lecture:

To top it off, I imagined them migrating to the tune of the Adventure Line music from The Stanley Parable (which, by the way, is also my ringtone. So now you know who to glare at if it goes off in lectures).
There! Now you're never going to be able to forget this, even when you desperately want to! Muahahaha!
Limb Development
We didn't really cover limb development in that much detail (in fact I'm fairly sure the lecturer pretty much skipped over the slide relating to limb development as we were going overtime), so all I can really tell you are the basics. Limbs develop from the somatopleure, which as I've mentioned several times, is the layer of the lateral plate mesoderm that associates with the body wall. The muscle and skin in this area is from the hypomere and is therefore supplied by ventral rami. Limb development starts from little limb buds that begin to form in the 4th week, which induce the proliferation of underlying mesoderm to create the limb. Formation of limbs are proximodistal (i.e. from the "close" end of the limb to the "far" end).
Early Embryology: The First Four Weeks
I have a test on fetal anatomy next Wednesday, which I'm worried about, because it seems like there's so much to learn! (Although let's be honest here- I have moderate test anxiety and I'd be worried even if the test was on my ability to count to 10.) Thus begins my anatomy blog posts! (Warning: they're probably going to be shitty due to lack of images, or images pulled from other sources. Pictures are kinda important for anatomy.)
Explain the process of fertilization and implantation
Identify main steps in blastocyst formation
I've actually written about all of this before in old posts about reproduction and pregnancy, but I didn't have as much of an understanding back then and thus my writing wasn't really that clear so I'm going to attempt again. Can't guarantee that my writing will be any clearer this time, though, but at least the concepts should be clearer in my own head so at least one person should benefit from this. Whoop-de-doop.
Anyway. Fertilisation. I'm sure you all know the general process that has to take place for sperm to get anywhere near an egg. (If you don't, then I guess that you'll have to redo year 8 sex ed.) At this stage the egg is surrounded by a couple of layers: a hard shell called the zona pellucida, and a layer of cells called the corona radiata. Part of the reason why millions of sperm need to be ejaculated is because no single sperm has enough enzymes to break through all of the layers. Firstly, the sperm release hyaluronidase which breaks down the hyaluronic acid holding together the cells of the corona radiata. Sperm then latch onto ZP2 glycoproteins in the zona pellucida and break through using zona-digestive enzymes located in the acrosome of the sperm (it's like a modified lysosome). After this is done, the first sperm to get through fuses with the membrane of the oocyte. A couple of processes then occur to stop more sperm from binding, otherwise you would end up with shitloads of chromosomes and that would be bad.
Oh and don't worry, I don't think you need to remember all of the details of how the sperm breaks through those layers. I just added them in for funsies. (Yes, I know, I have a weird idea of fun. Shut up.)
After the sperm fuses with the oocyte, a few more things happen- the female and male pronuclei (i.e. the nuclei of the egg and sperm, respectively) break down to release the chromosomes and then mitosis begins. The mitosis that happens at this stage is known as "cleavage," and involves many divisions but no cell growth in between. The cells are known as "blastomeres" and by 4 days there are around 32 of them that form a ball. This ball is known as a "morula."
Cells at the morula stage could theoretically keep dividing, but then they'd be so small it'd be absurd. Besides, you and I clearly aren't just tiny balls of cells. Something had to have happened to make us bigger.
That something is a process known as "hatching." Basically, the embryo "hatches" out of the zona pellucida. It happens around the time the morula enters the uterus, and to my understanding it's partly due to fluid seeping through the zona pellucida and into the intercellular spaces.
This fluid that seeps through forms a fluid-filled space known as the blastocystic cavity, or blastocoele. Of the remaining cells, some form a thin layer around the outside- these cells are known as trophoblasts and eventually form the placenta. The rest of the cells form a big mass which is why they're known as the "inner cell mass." The inner cell mass later becomes the embryo.
At around the end of the first week, implantation starts to occur. Firstly, the blastocyst fuses with the uterine wall. The trophoblasts then begin proliferating and differentiating (you'll find that "proliferating" and "differentiating" are pretty much the buzzwords of embryology). They differentiate into two layers: the cytotrophoblast which is pretty much just one layer of trophoblastic cells, and the syncytiotrophoblast which surrounds the cytotrophoblast and is made up of many cells fused together into one big one with several nuclei. The syncytiotrophoblast is the one that does the actual burrowing, while the cytotrophoblast maintains these cells. Blood vessels and so forth eventually start to form near the syncytiotrophoblast in order to allow for exchange of nutrients and waste products between the mother and the baby.
Outline the process of inner cell mass differentiation
The inner cell mass initially begins to differentiate into just two layers: the epiblast (upper layer) and the hypoblast (lower layer). Cavities begin to form within these layers. The cavity that forms within the epiblast is called the amnion, or amniotic cavity, whereas the cavity that forms within the hypoblast is known as the yolk sac. The area where the two layers come into contact is the "bilaminar embryonic disc."
A new layer of cells, the extraembryonic mesoderm, soon begin to form and line the rest of the cavity. Coelom (spaces) form between the cells of the extraembryonic mesoderm. These coelomic spaces then join up to form a large cavity called the chorionic cavity.
Thus, by the end of the week, you have a definite two-layer (bilaminar) embryo. There are a few more things that happen- like a secondary yolk sac forms and displaces the primary one (which eventually degenerates) and part of the hypoblast thickens to form the prechordal plate, which eventually becomes the cranial region of the embryo. (Cranial = head end, caudal = tail end.) Main point is, though, is that when the embryo is two weeks old, it's a two-layered disc.
Describe the process of differentiation of the basic germ layers
In week 2, the embryo has two layers; in week 3, the embryo will have three. (Nope, it doesn't continue beyond there: a 4-week old embryo does not, in fact, have four layers.) The process of the third layer forming is known as gastrulation.
Gastrulation begins with the formation of the primitive streak. The primitive streak is a thickening of the epiblast along the midline of the embryonic disc. It consists of a "primitive groove" with a "primitive node" at the end. The primitive node contains a depression known as the "primitive pit." It is this primitive streak that defines the cranial and caudal sides of the embryo.
For gastrulation to occur, epiblast cells near the primitive streak must first proliferate and differentiate (yes, it's those buzzwords again). These cells then migrate through the primitive streak in a process known as ingression. At first, the epiblast cells push away the hypoblast cells, replacing them with new endodermal cells to form the endoderm (endo = inside). Once the endoderm is done, the next lot of cells to migrate through become the mesoderm (meso = middle). That leaves the epiblast to become ectoderm (to my understanding it gets called "ectoderm" once the mesoderm is established).
Also of great importance during this time is the formation of the notochord. The notochord is a hollow tube that is responsible for signalling in the early embryo, and is thus important for signalling cells to proliferate and differentiate. The notochord extends cranially from the primitive node upwards to the prechordal plate (which, as I mentioned in the previous section, is a thickening of the hypoblast at the cranial end).
Formation of the notochord starts by prenotochordal cells migrating through the primitive streak and fusing with the endoderm to form the notochordal plate. Eventually this fused layer degenerates to form the definitive notochord.
Yet another important event that happens towards the end of the third week is the further development of the mesoderm. The mesoderm has three main sections. Paraxial mesoderm (i.e. "along the axis") lies next to the notochord. It is further differentiated into three types of clumps, called somites. Sclerotomes are responsible for forming the skeleton, myotomes are responsible for forming voluntary muscle and dermatomes are responsible for forming the dermis of the skin. Lateral to the paraxial mesoderm is the intermediate mesoderm, which is responsible for the formation of the kidneys, adrenal cortex and genitals.
The third and final section of the mesoderm is the lateral plate mesoderm. During development, spaces begin to form within the lateral plate mesoderm, effectively splitting it into two layers with an intraembryonic coelom in between (contrast with the extraembryonic coelom, which is a space that lies outside the embryo). One layer associates with the ectoderm and is known as the somatopleure. The other layer associates with the endoderm and is known as the visceropleure.
A final important development at this time is that the cardiovascular system begins to form. Formation of the cardiovascular system starts off with blood vessels appearing in the yolk sac, and then in the embryo itself. A tubular heart also forms at this time.
During week four of development, no new layers are created, but the layers that are present begin to grow. As they grow, they begin to fold on themselves so that the embryo finally looks less like a disc and more like an embryo. Two types of folding occur simultaneously. One is craniocaudal (i.e. "head to tail") folding, caused by rapid growth of the ectoderm, while the other type is lateral folding, caused by rapid growth of the mesoderm. Together, they result in a "tube within a tube" body plan, where there is an outer tube that represents the general outline of the embryo, and an inner tube that is the gut of the embryo.
Explain processes associated with the establishment of the neural tube
The process in which the neural tube develops is known as neurulation. In neurulation, ectoderm cranial to the primitive node begins to thicken to form the neural plate. The edges of the neural plate begin to fold up to create neural folds, while the bit in between becomes the neural groove. The neural folds continue to rise until they meet each other, forming the neural tube in the middle. After the neural folds meet and fuse, some of the cells break off to form neural crest cells- these cells become the nerves of the peripheral nervous system, among other things, as highlighted in the list below.
The meeting of the neural folds begins in the neck, and then proceeds to "zip up" in both directions (cranial and caudal). The cranial end that isn't zipped up yet is called the cranial neuropore, while the caudal end that isn't zipped up yet is the caudal neuropore. The cranial end closes before the caudal end. Failure to close can result in a serious condition called spina bifida. The risk of spina bifida can be reduced by taking folic acid during pregnancy.
Identify the major primordia associated with the germ layers
List time!
Ectoderm
Explain the process of fertilization and implantation
Identify main steps in blastocyst formation
I've actually written about all of this before in old posts about reproduction and pregnancy, but I didn't have as much of an understanding back then and thus my writing wasn't really that clear so I'm going to attempt again. Can't guarantee that my writing will be any clearer this time, though, but at least the concepts should be clearer in my own head so at least one person should benefit from this. Whoop-de-doop.
Anyway. Fertilisation. I'm sure you all know the general process that has to take place for sperm to get anywhere near an egg. (If you don't, then I guess that you'll have to redo year 8 sex ed.) At this stage the egg is surrounded by a couple of layers: a hard shell called the zona pellucida, and a layer of cells called the corona radiata. Part of the reason why millions of sperm need to be ejaculated is because no single sperm has enough enzymes to break through all of the layers. Firstly, the sperm release hyaluronidase which breaks down the hyaluronic acid holding together the cells of the corona radiata. Sperm then latch onto ZP2 glycoproteins in the zona pellucida and break through using zona-digestive enzymes located in the acrosome of the sperm (it's like a modified lysosome). After this is done, the first sperm to get through fuses with the membrane of the oocyte. A couple of processes then occur to stop more sperm from binding, otherwise you would end up with shitloads of chromosomes and that would be bad.
Oh and don't worry, I don't think you need to remember all of the details of how the sperm breaks through those layers. I just added them in for funsies. (Yes, I know, I have a weird idea of fun. Shut up.)
After the sperm fuses with the oocyte, a few more things happen- the female and male pronuclei (i.e. the nuclei of the egg and sperm, respectively) break down to release the chromosomes and then mitosis begins. The mitosis that happens at this stage is known as "cleavage," and involves many divisions but no cell growth in between. The cells are known as "blastomeres" and by 4 days there are around 32 of them that form a ball. This ball is known as a "morula."
Cells at the morula stage could theoretically keep dividing, but then they'd be so small it'd be absurd. Besides, you and I clearly aren't just tiny balls of cells. Something had to have happened to make us bigger.
That something is a process known as "hatching." Basically, the embryo "hatches" out of the zona pellucida. It happens around the time the morula enters the uterus, and to my understanding it's partly due to fluid seeping through the zona pellucida and into the intercellular spaces.
This fluid that seeps through forms a fluid-filled space known as the blastocystic cavity, or blastocoele. Of the remaining cells, some form a thin layer around the outside- these cells are known as trophoblasts and eventually form the placenta. The rest of the cells form a big mass which is why they're known as the "inner cell mass." The inner cell mass later becomes the embryo.
At around the end of the first week, implantation starts to occur. Firstly, the blastocyst fuses with the uterine wall. The trophoblasts then begin proliferating and differentiating (you'll find that "proliferating" and "differentiating" are pretty much the buzzwords of embryology). They differentiate into two layers: the cytotrophoblast which is pretty much just one layer of trophoblastic cells, and the syncytiotrophoblast which surrounds the cytotrophoblast and is made up of many cells fused together into one big one with several nuclei. The syncytiotrophoblast is the one that does the actual burrowing, while the cytotrophoblast maintains these cells. Blood vessels and so forth eventually start to form near the syncytiotrophoblast in order to allow for exchange of nutrients and waste products between the mother and the baby.
Outline the process of inner cell mass differentiation
The inner cell mass initially begins to differentiate into just two layers: the epiblast (upper layer) and the hypoblast (lower layer). Cavities begin to form within these layers. The cavity that forms within the epiblast is called the amnion, or amniotic cavity, whereas the cavity that forms within the hypoblast is known as the yolk sac. The area where the two layers come into contact is the "bilaminar embryonic disc."
A new layer of cells, the extraembryonic mesoderm, soon begin to form and line the rest of the cavity. Coelom (spaces) form between the cells of the extraembryonic mesoderm. These coelomic spaces then join up to form a large cavity called the chorionic cavity.
Thus, by the end of the week, you have a definite two-layer (bilaminar) embryo. There are a few more things that happen- like a secondary yolk sac forms and displaces the primary one (which eventually degenerates) and part of the hypoblast thickens to form the prechordal plate, which eventually becomes the cranial region of the embryo. (Cranial = head end, caudal = tail end.) Main point is, though, is that when the embryo is two weeks old, it's a two-layered disc.
Describe the process of differentiation of the basic germ layers
In week 2, the embryo has two layers; in week 3, the embryo will have three. (Nope, it doesn't continue beyond there: a 4-week old embryo does not, in fact, have four layers.) The process of the third layer forming is known as gastrulation.
Gastrulation begins with the formation of the primitive streak. The primitive streak is a thickening of the epiblast along the midline of the embryonic disc. It consists of a "primitive groove" with a "primitive node" at the end. The primitive node contains a depression known as the "primitive pit." It is this primitive streak that defines the cranial and caudal sides of the embryo.
For gastrulation to occur, epiblast cells near the primitive streak must first proliferate and differentiate (yes, it's those buzzwords again). These cells then migrate through the primitive streak in a process known as ingression. At first, the epiblast cells push away the hypoblast cells, replacing them with new endodermal cells to form the endoderm (endo = inside). Once the endoderm is done, the next lot of cells to migrate through become the mesoderm (meso = middle). That leaves the epiblast to become ectoderm (to my understanding it gets called "ectoderm" once the mesoderm is established).
Also of great importance during this time is the formation of the notochord. The notochord is a hollow tube that is responsible for signalling in the early embryo, and is thus important for signalling cells to proliferate and differentiate. The notochord extends cranially from the primitive node upwards to the prechordal plate (which, as I mentioned in the previous section, is a thickening of the hypoblast at the cranial end).
Formation of the notochord starts by prenotochordal cells migrating through the primitive streak and fusing with the endoderm to form the notochordal plate. Eventually this fused layer degenerates to form the definitive notochord.
Yet another important event that happens towards the end of the third week is the further development of the mesoderm. The mesoderm has three main sections. Paraxial mesoderm (i.e. "along the axis") lies next to the notochord. It is further differentiated into three types of clumps, called somites. Sclerotomes are responsible for forming the skeleton, myotomes are responsible for forming voluntary muscle and dermatomes are responsible for forming the dermis of the skin. Lateral to the paraxial mesoderm is the intermediate mesoderm, which is responsible for the formation of the kidneys, adrenal cortex and genitals.
The third and final section of the mesoderm is the lateral plate mesoderm. During development, spaces begin to form within the lateral plate mesoderm, effectively splitting it into two layers with an intraembryonic coelom in between (contrast with the extraembryonic coelom, which is a space that lies outside the embryo). One layer associates with the ectoderm and is known as the somatopleure. The other layer associates with the endoderm and is known as the visceropleure.
A final important development at this time is that the cardiovascular system begins to form. Formation of the cardiovascular system starts off with blood vessels appearing in the yolk sac, and then in the embryo itself. A tubular heart also forms at this time.
During week four of development, no new layers are created, but the layers that are present begin to grow. As they grow, they begin to fold on themselves so that the embryo finally looks less like a disc and more like an embryo. Two types of folding occur simultaneously. One is craniocaudal (i.e. "head to tail") folding, caused by rapid growth of the ectoderm, while the other type is lateral folding, caused by rapid growth of the mesoderm. Together, they result in a "tube within a tube" body plan, where there is an outer tube that represents the general outline of the embryo, and an inner tube that is the gut of the embryo.
Explain processes associated with the establishment of the neural tube
The process in which the neural tube develops is known as neurulation. In neurulation, ectoderm cranial to the primitive node begins to thicken to form the neural plate. The edges of the neural plate begin to fold up to create neural folds, while the bit in between becomes the neural groove. The neural folds continue to rise until they meet each other, forming the neural tube in the middle. After the neural folds meet and fuse, some of the cells break off to form neural crest cells- these cells become the nerves of the peripheral nervous system, among other things, as highlighted in the list below.
The meeting of the neural folds begins in the neck, and then proceeds to "zip up" in both directions (cranial and caudal). The cranial end that isn't zipped up yet is called the cranial neuropore, while the caudal end that isn't zipped up yet is the caudal neuropore. The cranial end closes before the caudal end. Failure to close can result in a serious condition called spina bifida. The risk of spina bifida can be reduced by taking folic acid during pregnancy.
Identify the major primordia associated with the germ layers
List time!
Ectoderm
- Surface ectoderm- epidermis of the skin and epidermal appendages (i.e. hair, nails, etc.), anterior pituitary gland, roof of oral cavity, enamel
- Neural tube ectoderm- central nervous system
- Neural crest ectoderm- sensory epithelia of the eyes, ears and nose, peripheral nervous system, adrenal medulla
Mesoderm
- Paraxial mesoderm (somites)- voluntary muscle (from myotome), axial skeleton (sclerotome), dermis of skin (dermatome)
- Intermediate mesoderm- gonads, kidneys, adrenal cortex
- Lateral plate mesoderm- contributes to the serous membranes lining body cavities (pericardial, pleural, peritoneal)
Endoderm
- Epithelial lining of gastrointestinal and respiratory tracts, urinary bladder and urethra etc.
- Thyroid and parathyroid glands
- Thymus, liver and pancreas
Whew! That was long...
Saturday, March 19, 2016
Purifying Proteins
In earlier posts I've talked about how to replicate and amplify DNA sequences so that you can make shitloads of proteins. Well, I've realised that I haven't actually spoken about how to extract and purify all of those proteins that you just made (or bullied a cell into making for you).
A lot of these purification processes actually require that you've made a purifiable protein to begin with. For example, with a His tag, you need to add some DNA coding for a bunch of histidine residues to the beginning or end of the sequence. I've actually spoken about His tags in an earlier post: Introduction to Cloning. Essentially the proteins are washed down a column, and the ones with His tags bind to nickel beads in the column. Eventually all that's left in the column are the His-tagged proteins bound to metal beads. These can then be eluted from the column by lowering the pH or by adding extra histidine or imidazole.
A second type of purification process is called GST-Fusion, which of course has absolutely nothing to do with Goods and Services Taxes. GST stands for glutathione-S-transferase. DNA coding for glutathione-S-transferase is positioned right before the DNA for the protein of interest, so that the protein is attached to glutathione-S-transferase. Glutathione-S-transferase attaches to glutathione (a.k.a. GSH), rather than nickel beads. It can be eluted with free glutathione. The GST part can then be removed by cleavage with Xa protease at a site which, like the GST enzyme itself, has also been engineered into the protein.
A lot of these purification processes actually require that you've made a purifiable protein to begin with. For example, with a His tag, you need to add some DNA coding for a bunch of histidine residues to the beginning or end of the sequence. I've actually spoken about His tags in an earlier post: Introduction to Cloning. Essentially the proteins are washed down a column, and the ones with His tags bind to nickel beads in the column. Eventually all that's left in the column are the His-tagged proteins bound to metal beads. These can then be eluted from the column by lowering the pH or by adding extra histidine or imidazole.
A second type of purification process is called GST-Fusion, which of course has absolutely nothing to do with Goods and Services Taxes. GST stands for glutathione-S-transferase. DNA coding for glutathione-S-transferase is positioned right before the DNA for the protein of interest, so that the protein is attached to glutathione-S-transferase. Glutathione-S-transferase attaches to glutathione (a.k.a. GSH), rather than nickel beads. It can be eluted with free glutathione. The GST part can then be removed by cleavage with Xa protease at a site which, like the GST enzyme itself, has also been engineered into the protein.
DNA Sequencing and Synthesis
Last post on recombinant DNA technology for this unit! (That went quickly...)
Understand the principle of dideoxy sequencing.
Dideoxy sequencing, also known as the Sanger method after the guy who invented it, is a pretty reliable technique for sequencing DNA.
In traditional dideoxy sequencing, you need the following materials:
Understand how PCR can be used in the dideoxy sequencing reaction and how the sequencing is automated.
PCR can be used in the first part of the sequencing reaction (i.e. the bit with the synthesising of fragments ending in dideoxynucleotides) in a technique known as "cycle sequencing," or "linear amplification sequencing." In this process, only one strand is primed so that a linear (rather than exponential) amplification of products is achieved- hence "linear amplification sequencing."
In automated sequencing, the process is carried out in a single tube (rather than four tubes for the four different dideoxynucleotides). This is achieved by fluorescently labelling the ddNTPs (sorry, got sick of typing out "dideoxynucleotides") with different colours. After running the fragments through electrophoresis (only one lane required this time), a machine can detect the different wavelengths of emitted light to give a reading of the sequence.
Be familiar with new and rapid “next generation sequencing”.
Nowadays, there are faster and cheaper methods of sequencing DNA, though they are not necessarily as accurate as the dideoxy/Sanger method outlined above. Most forms of "next generation sequencing" involve creating billions of tiny fragments of DNA (30-70 base pairs long) which are immobilised to beads or chips. This immobilisation is done by adding an oligo-dA tail to the fragments, which bind to oligo-dT molecules in the beads or chips.
Fluorescently labelled dNTPs (not ddNTPs) are used for "next generation sequencing." They will bind to oligo-dT anchors if the fragment next to it has a complementary base. This will then cause that strand to fluoresce. The fluorescent tag is then removed, and a different fluorescently labelled dNTP is added. The fluorescence can be picked up by a computer to sequence the DNA.
Be familiar with how DNA can be chemically synthesized and the uses of synthetic DNA.
One thing you might have wondered throughout all of this is how primers and so forth are synthesised. One method of synthesising oligonucleotides is the phosphoramidite procedure, which synthesises oligonucleotides from 3' to 5' (opposite of the normal direction).
The phosphoramidite procedure starts by anchoring a nucleoside to a glass support at the 3' end. This nucleoside also has a blocking group (usually dimethyoxytrityl, a.k.a. DMTr) attached to the 5' end to prevent spontaneous reaction. Throughout this procedure, most of the bases added also have blocking groups added to them until they are washed off at the end.
The first step of the main part of the procedure is washing off the DMTr group by using trichloroacetic acid. Next the second base is added, except it's not added in the form of a nucleotide: it's added in the form of a nucleoside phosphoramidite derivative. This nucleoside phosphoramidite derivative is essentially just a nucleoside with a DMTr blocking group attached to the 5' end and a phosphoramidite group attached to the 3' end. Phosphoramidite groups centre around a trivalent phosphorus (i.e. a phosphorus atom that only forms 3 bonds rather than 5 like in phosphate). They also have other crap attached to them but I'm not going to go into that because it's not important. What's important is that this phosphoramidite group can be activated by a weak acid, such as tetrazole, allowing it to rapidly react with the 5' end of the first nucleotide. Aqueous iodine is then added to form a stable phosphate group. These steps are then repeated until the desired chain length is reached.
At the end of the process, the oligonucleotide is cleaved from the support by using ammonium hydroxide, which also removes blocking groups from the bases.
We're now done for this topic! Whew! (I might make a short mention on His tagging and GST-Fusion tagging in a later post, since that was covered in an earlier lecture but not in my posts here.)
Understand the principle of dideoxy sequencing.
Dideoxy sequencing, also known as the Sanger method after the guy who invented it, is a pretty reliable technique for sequencing DNA.
In traditional dideoxy sequencing, you need the following materials:
- Template strands for the DNA that you want to sequence
- A primer
- DNA Polymerase I
- All four nucleotides- at least one of these should be attached to a radioactive phosphate
- Dideoxynucleotides. These are nucleotides with two -H groups as opposed to -OH groups (hence di- and deoxy-). The other H group is on the 3' carbon. Yup, that means that there's no 3'-OH, which means that new nucleotides cannot be added after the addition of a dideoxynucleotide. This is pretty important, as we're about to see.
Four reactions need to be carried out, each with a different dideoxynucleotide (i.e. one reaction needs to be carried out with dideoxyadenosine, one with dideoxythymidine etc.). Essentially, in each reaction, DNA Polymerase I is used to synthesise a new strand, but it will terminate whenever a dideoxynucleotide is added. (The concentration of dideoxynucleotides is kept reasonably low so that the chain won't always stop at the first incidence of whatever base you're looking at.)
The fragments created from the previous step are then run through an electrophoresis process. Fragments from each reaction are run through different lanes so that you can keep track of which fragments end with dideoxyadenosine, which fragments end with dideoxycytidine etc. The gel used is usually 4-6% polyacrylamide and 6M urea (the urea is used in order to denature the DNA so that its secondary structure won't interfere with electrophoresis). Like in normal electrophoresis, smaller fragments migrate through the gel faster. Thus by reading "up" the gel you can see what the sequence of bases are.
Understand how PCR can be used in the dideoxy sequencing reaction and how the sequencing is automated.
PCR can be used in the first part of the sequencing reaction (i.e. the bit with the synthesising of fragments ending in dideoxynucleotides) in a technique known as "cycle sequencing," or "linear amplification sequencing." In this process, only one strand is primed so that a linear (rather than exponential) amplification of products is achieved- hence "linear amplification sequencing."
In automated sequencing, the process is carried out in a single tube (rather than four tubes for the four different dideoxynucleotides). This is achieved by fluorescently labelling the ddNTPs (sorry, got sick of typing out "dideoxynucleotides") with different colours. After running the fragments through electrophoresis (only one lane required this time), a machine can detect the different wavelengths of emitted light to give a reading of the sequence.
Be familiar with new and rapid “next generation sequencing”.
Nowadays, there are faster and cheaper methods of sequencing DNA, though they are not necessarily as accurate as the dideoxy/Sanger method outlined above. Most forms of "next generation sequencing" involve creating billions of tiny fragments of DNA (30-70 base pairs long) which are immobilised to beads or chips. This immobilisation is done by adding an oligo-dA tail to the fragments, which bind to oligo-dT molecules in the beads or chips.
Fluorescently labelled dNTPs (not ddNTPs) are used for "next generation sequencing." They will bind to oligo-dT anchors if the fragment next to it has a complementary base. This will then cause that strand to fluoresce. The fluorescent tag is then removed, and a different fluorescently labelled dNTP is added. The fluorescence can be picked up by a computer to sequence the DNA.
Be familiar with how DNA can be chemically synthesized and the uses of synthetic DNA.
One thing you might have wondered throughout all of this is how primers and so forth are synthesised. One method of synthesising oligonucleotides is the phosphoramidite procedure, which synthesises oligonucleotides from 3' to 5' (opposite of the normal direction).
The phosphoramidite procedure starts by anchoring a nucleoside to a glass support at the 3' end. This nucleoside also has a blocking group (usually dimethyoxytrityl, a.k.a. DMTr) attached to the 5' end to prevent spontaneous reaction. Throughout this procedure, most of the bases added also have blocking groups added to them until they are washed off at the end.
The first step of the main part of the procedure is washing off the DMTr group by using trichloroacetic acid. Next the second base is added, except it's not added in the form of a nucleotide: it's added in the form of a nucleoside phosphoramidite derivative. This nucleoside phosphoramidite derivative is essentially just a nucleoside with a DMTr blocking group attached to the 5' end and a phosphoramidite group attached to the 3' end. Phosphoramidite groups centre around a trivalent phosphorus (i.e. a phosphorus atom that only forms 3 bonds rather than 5 like in phosphate). They also have other crap attached to them but I'm not going to go into that because it's not important. What's important is that this phosphoramidite group can be activated by a weak acid, such as tetrazole, allowing it to rapidly react with the 5' end of the first nucleotide. Aqueous iodine is then added to form a stable phosphate group. These steps are then repeated until the desired chain length is reached.
At the end of the process, the oligonucleotide is cleaved from the support by using ammonium hydroxide, which also removes blocking groups from the bases.
We're now done for this topic! Whew! (I might make a short mention on His tagging and GST-Fusion tagging in a later post, since that was covered in an earlier lecture but not in my posts here.)
Friday, March 18, 2016
Genetic Recombination and the Use of Recombinase in Cloning
I must admit that my friend and I spent part of the lecture playing Hangman, but hopefully the main details are fine :)
Be familiar with the ability of DNA ligase to join DNA molecules and how to overcome vector self-ligation.
Okay, you should be reasonably familiar with the idea that DNA ligase joins DNA molecules. If not, have another look at my post on enzymes involved in DNA replication and repair.
Now let's have a look at the idea of self-ligation. Self-ligation occurs when, instead of a fragment of DNA being inserted into a plasmid, the two cut ends of the plasmid simply snap back together (i.e. self-ligate). This can be easily prevented by using alkaline phosphatase to remove phosphate groups on the plasmids. Without these phosphate groups, the plasmid cannot self-ligate, but fragments can still be inserted. This does leave single-stranded nicks where the plasmid lacks phosphates, but the effect of this is negligible due to the size of the inserted fragment and the distance between the two nicks.
Be familiar with the types of site-specific recombination.
There are three main types of site-specific recombination: insertion, deletion or inversion. They're pretty much self-explanatory: insertion involves the insertion of a new DNA fragment, deletion involves its deletion, and inversion involves re-inserting it backwards (i.e. inverting it). These reactions are catalysed by recombinases, which are part of a larger family of enzymes known as integrases. There are two essential parts of the DNA itself that facilitate site-specific recombination: recognition sites for the recombinases, and a crossover region where cutting and rejoining occurs. The crossover region also confers directionality, which is important in the case of inversion.
Understand the mechanism of action of Cre recombinase.
Cre recombinase is a recombinase (duh) taken from Phage P1. Its original function is to circularise the phage genome when it infects bacteria. Cre recombinase recognises a certain site called the LoxP site. The LoxP site contains an 8 base pair core sequence which is not palindromic and therefore confers directionality. This 8 base pair core sequence is flanked by two palindromic 13 base pair sequences. (By palindromic, I mean that the one before the core sequence reads the same as the one after, but backwards.)
Cre recombinase is made up of four identical subunits, each of which has a tyrosine in the active site. This tyrosine breaks the linkage between the 5'-OH and the phosphate of a nucleotide, forming a covalent intermediate until another nucleotide comes along. There are other recombinases that have serine in the active site- these ones break the linkage between the 3'-OH and the phosphate of the next nucleotide. The formation of these intermediates eliminates the need for ATP or other energy sources.
Cre recombinase works by cleaving and rejoining two strands simultaneously. Subunits R1 and R2 hold onto one DNA molecule at the palindromic sequences while R3 and R4 hold onto the other (again, at the palindromic sequences).
One of the benefits of using Cre recombinase is that its recognition sequence, LoxP, doesn't occur naturally in plants or animals. Hence, engineering a LoxP site into the genome and then using Cre recombinase will not cause unwanted cuts. (Of course, there's probably a tiny chance that there could be the exact same site somewhere in the genome by chance, but that chance is really, really negligible at best.)
Be familiar with applications of Cre recombinase and LoxP sites in cloning and conditional gene targeting.
As I just mentioned, Cre recombinase and LoxP are good for "cutting and pasting" (similar to restriction enzymes) as LoxP sites don't occur naturally in most plants and animals. Hence they are good for modifying and moving around genes. LoxP sites can also be used for conditional gene targeting- the so-called "knocking out" of genes.
I'm now going attempt to explain conditional gene targeting, using mice as an example. To create mice that have a gene "knocked out," you first need to start with two kinds of transgenic mice. One has the gene for Cre recombinase tied to an inducible and tissue-specific promoter, whereas the other kind has loxP sites flanking one of the exons of the gene in question. Following breeding, some of the mice should have both characteristics: loxP sites and the gene for Cre recombinase. The gene that you are studying can then be "knocked out" by inducing the production of Cre recombinase. This allows more control over timing of gene expression, and also allows you to see the effects of genes that could have been lethal during embryonic development.
Be familiar with the ability of DNA ligase to join DNA molecules and how to overcome vector self-ligation.
Okay, you should be reasonably familiar with the idea that DNA ligase joins DNA molecules. If not, have another look at my post on enzymes involved in DNA replication and repair.
Now let's have a look at the idea of self-ligation. Self-ligation occurs when, instead of a fragment of DNA being inserted into a plasmid, the two cut ends of the plasmid simply snap back together (i.e. self-ligate). This can be easily prevented by using alkaline phosphatase to remove phosphate groups on the plasmids. Without these phosphate groups, the plasmid cannot self-ligate, but fragments can still be inserted. This does leave single-stranded nicks where the plasmid lacks phosphates, but the effect of this is negligible due to the size of the inserted fragment and the distance between the two nicks.
Be familiar with the types of site-specific recombination.
There are three main types of site-specific recombination: insertion, deletion or inversion. They're pretty much self-explanatory: insertion involves the insertion of a new DNA fragment, deletion involves its deletion, and inversion involves re-inserting it backwards (i.e. inverting it). These reactions are catalysed by recombinases, which are part of a larger family of enzymes known as integrases. There are two essential parts of the DNA itself that facilitate site-specific recombination: recognition sites for the recombinases, and a crossover region where cutting and rejoining occurs. The crossover region also confers directionality, which is important in the case of inversion.
Understand the mechanism of action of Cre recombinase.
Cre recombinase is a recombinase (duh) taken from Phage P1. Its original function is to circularise the phage genome when it infects bacteria. Cre recombinase recognises a certain site called the LoxP site. The LoxP site contains an 8 base pair core sequence which is not palindromic and therefore confers directionality. This 8 base pair core sequence is flanked by two palindromic 13 base pair sequences. (By palindromic, I mean that the one before the core sequence reads the same as the one after, but backwards.)
Cre recombinase is made up of four identical subunits, each of which has a tyrosine in the active site. This tyrosine breaks the linkage between the 5'-OH and the phosphate of a nucleotide, forming a covalent intermediate until another nucleotide comes along. There are other recombinases that have serine in the active site- these ones break the linkage between the 3'-OH and the phosphate of the next nucleotide. The formation of these intermediates eliminates the need for ATP or other energy sources.
Cre recombinase works by cleaving and rejoining two strands simultaneously. Subunits R1 and R2 hold onto one DNA molecule at the palindromic sequences while R3 and R4 hold onto the other (again, at the palindromic sequences).
One of the benefits of using Cre recombinase is that its recognition sequence, LoxP, doesn't occur naturally in plants or animals. Hence, engineering a LoxP site into the genome and then using Cre recombinase will not cause unwanted cuts. (Of course, there's probably a tiny chance that there could be the exact same site somewhere in the genome by chance, but that chance is really, really negligible at best.)
Be familiar with applications of Cre recombinase and LoxP sites in cloning and conditional gene targeting.
As I just mentioned, Cre recombinase and LoxP are good for "cutting and pasting" (similar to restriction enzymes) as LoxP sites don't occur naturally in most plants and animals. Hence they are good for modifying and moving around genes. LoxP sites can also be used for conditional gene targeting- the so-called "knocking out" of genes.
I'm now going attempt to explain conditional gene targeting, using mice as an example. To create mice that have a gene "knocked out," you first need to start with two kinds of transgenic mice. One has the gene for Cre recombinase tied to an inducible and tissue-specific promoter, whereas the other kind has loxP sites flanking one of the exons of the gene in question. Following breeding, some of the mice should have both characteristics: loxP sites and the gene for Cre recombinase. The gene that you are studying can then be "knocked out" by inducing the production of Cre recombinase. This allows more control over timing of gene expression, and also allows you to see the effects of genes that could have been lethal during embryonic development.
Cloning by PCR and Mutagenesis
Appreciate that there are different
approaches to the cloning of specific
genes.
A moment of silence here while we appreciate all the different approaches...
Okay, back to being serious. There's really just two main approaches (well, at least only two that we learned about during the lecture). They both start off with figuring out what protein you want to clone, determining some of the N-terminal amino acid sequence and from there taking a stab at what the potential DNA sequence would be (presumably taking into account that some amino acids can be coded for by more than one codon).
From there you can take one of two approaches, depending on what technology is available to you. Since several different genomes have already been sequenced, the easiest approach nowadays would generally be to search the database for the gene of interest. You might also be able to find similar genes coding for other proteins, and from there you might be able to get some kind of idea of what your protein does. To be a bit more certain, though, you can amplify the gene (and therefore protein production), allowing you to study the protein more and find out more about it. To amplify the gene, use the database to help you design some oligonucleotide primers so that you can use PCR (more on PCR in an earlier post). This amplified DNA can then be cloned into an expression vector (see the end of my most recent post) to create lots of protein.
An alternative approach can be used if you don't have access to a database for searching for genes. After working out a potential DNA sequence, you can make a DNA probe using this sequence data. This probe can then be used to find the gene of interest in a gene library (more on gene libraries in a bit). Once you've found your gene, you can clone it and so forth.
Be familiar with cDNA library construction and traditional screening by hybridisation.
cDNA libraries can be bought commercially, but just in case you wanted to make your own, I'm gonna tell you how (or rather, the theory behind it anyway). *Do not try this at home*
cDNA is essentially the complement of mRNA. To create cDNA, simply use the enzyme reverse transcriptase to transcribe the mRNA into DNA. Remove the mRNA with alkali and add a poly-G tail (this is so you can add a poly-C primer for synthesis of the other strand). Synthesise the other strand using DNA polymerase.
Next you have to do a couple more fancy things with the cDNA. Firstly, you have to methylate it so that restriction enzymes won't cut it where you don't want it to be cut. Secondly, you have to add EcoRI linkers to either side (essentially the "cutting sites" for EcoRI). Cleave them with EcoRI to form sticky ends.
Now for the recombination part! Take some bacteriophage lambda (a bacteriophage is essentially a virus that infects bacteria) and cleave its DNA with EcoRI. Ligate this to the cDNA. Now you can package this cDNA into bacteriophage lambda so that it can go ahead and infect EcoRI with some shiny new cDNA!
cDNA libraries can be used to screen by hybridisation of a probe, as alluded to in the previous section. Firstly, the library has to be plated out and a nitrocellulose membrane placed on top. The colonies or plaques will be transferred to this membrane due to the binding of DNA. Place the nitrocellulose membrane in a plastic bag along with a solution containing a radioactive probe (I don't know how long for, sorry :P). Later wash and radiograph the nitrocellulose paper to find the location of the radioactive probes, which should be located in the same place as colonies containing the gene of interest. These colonies can then be removed from the agar plate and cultured in nutrient broths.
Understand the principle of PCR.
Know how PCR can be used for the cloning of specific genes.
See my previous post on PCR.
Be familiar with how PCR can be used for site-directed mutagenesis.
One way of testing out how a mutation affects the function of a protein is to create a primer with a mutation in it. The mutation can be located almost anywhere in a primer, except for the 3' end, because that has to fit to the DNA well for synthesis to proceed.
A moment of silence here while we appreciate all the different approaches...
Okay, back to being serious. There's really just two main approaches (well, at least only two that we learned about during the lecture). They both start off with figuring out what protein you want to clone, determining some of the N-terminal amino acid sequence and from there taking a stab at what the potential DNA sequence would be (presumably taking into account that some amino acids can be coded for by more than one codon).
From there you can take one of two approaches, depending on what technology is available to you. Since several different genomes have already been sequenced, the easiest approach nowadays would generally be to search the database for the gene of interest. You might also be able to find similar genes coding for other proteins, and from there you might be able to get some kind of idea of what your protein does. To be a bit more certain, though, you can amplify the gene (and therefore protein production), allowing you to study the protein more and find out more about it. To amplify the gene, use the database to help you design some oligonucleotide primers so that you can use PCR (more on PCR in an earlier post). This amplified DNA can then be cloned into an expression vector (see the end of my most recent post) to create lots of protein.
An alternative approach can be used if you don't have access to a database for searching for genes. After working out a potential DNA sequence, you can make a DNA probe using this sequence data. This probe can then be used to find the gene of interest in a gene library (more on gene libraries in a bit). Once you've found your gene, you can clone it and so forth.
Be familiar with cDNA library construction and traditional screening by hybridisation.
cDNA libraries can be bought commercially, but just in case you wanted to make your own, I'm gonna tell you how (or rather, the theory behind it anyway). *Do not try this at home*
cDNA is essentially the complement of mRNA. To create cDNA, simply use the enzyme reverse transcriptase to transcribe the mRNA into DNA. Remove the mRNA with alkali and add a poly-G tail (this is so you can add a poly-C primer for synthesis of the other strand). Synthesise the other strand using DNA polymerase.
Next you have to do a couple more fancy things with the cDNA. Firstly, you have to methylate it so that restriction enzymes won't cut it where you don't want it to be cut. Secondly, you have to add EcoRI linkers to either side (essentially the "cutting sites" for EcoRI). Cleave them with EcoRI to form sticky ends.
Now for the recombination part! Take some bacteriophage lambda (a bacteriophage is essentially a virus that infects bacteria) and cleave its DNA with EcoRI. Ligate this to the cDNA. Now you can package this cDNA into bacteriophage lambda so that it can go ahead and infect EcoRI with some shiny new cDNA!
cDNA libraries can be used to screen by hybridisation of a probe, as alluded to in the previous section. Firstly, the library has to be plated out and a nitrocellulose membrane placed on top. The colonies or plaques will be transferred to this membrane due to the binding of DNA. Place the nitrocellulose membrane in a plastic bag along with a solution containing a radioactive probe (I don't know how long for, sorry :P). Later wash and radiograph the nitrocellulose paper to find the location of the radioactive probes, which should be located in the same place as colonies containing the gene of interest. These colonies can then be removed from the agar plate and cultured in nutrient broths.
Understand the principle of PCR.
Know how PCR can be used for the cloning of specific genes.
See my previous post on PCR.
Be familiar with how PCR can be used for site-directed mutagenesis.
One way of testing out how a mutation affects the function of a protein is to create a primer with a mutation in it. The mutation can be located almost anywhere in a primer, except for the 3' end, because that has to fit to the DNA well for synthesis to proceed.
Cloning Vectors and Restriction Enzymes
So, it turns out that this online test is actually an in-class test next week. Which means I might actually have to pay attention to details and commit them to memory just in case they ask some horrible questions. Yikes. (I'm one of those people who does better at understanding general concepts rather than remembering details, so this will be fun.)
To understand the types of cuts to DNA made by restriction enzymes.
See my previous post- Introduction to Cloning.
To be familiar with some of the applications of restriction enzymes in recombinant DNA technology.
This is something that's probably going to crop up again and again over the next few posts in more detail. In a nutshell, though: recombinant DNA technology is all about "cutting and pasting" bits of DNA together. Restriction enzymes act as "scissors" to allow the cutting to be done. (DNA ligase acts as the paste.)
To be aware of the essential characteristics of plasmid vectors.
There are several different kinds of plasmid vectors, but they all have some essential features:
To be familiar with other cloning vectors including BACs and YACs.
BACs, or Bacterial Artificial Chromosomes, are plasmids that can be used to copy fairly large genomic fragments (100 000-300 000 base pairs). They are based on the F plasmid of E. coli (yeah, I get the feeling that we're going to be hearing a helluva lot about E. coli for the rest of my degree). Here are some of the essential characteristics in a list, because lists are great:
To know the important characteristics of expression vectors and how they can be used to express the protein encoded by a foreign gene.
Expression vectors are used to stimulate the production of a protein. The most important characteristic of an expression vector is that it has an inducible promoter (i.e. a promoter that you can switch on and off). For example, the lactose operon, which I've written about before, has a promoter which can be induced by allolactose (although in the lab IPTG, a lactose analogue, is more commonly used). In order to get this vector to produce something else, however, the rest of the gene is simply switched out with the gene of interest.
To understand the types of cuts to DNA made by restriction enzymes.
See my previous post- Introduction to Cloning.
To be familiar with some of the applications of restriction enzymes in recombinant DNA technology.
This is something that's probably going to crop up again and again over the next few posts in more detail. In a nutshell, though: recombinant DNA technology is all about "cutting and pasting" bits of DNA together. Restriction enzymes act as "scissors" to allow the cutting to be done. (DNA ligase acts as the paste.)
To be aware of the essential characteristics of plasmid vectors.
There are several different kinds of plasmid vectors, but they all have some essential features:
- An origin of replication (ori) site to allow the plasmid to be replicated.
- Genes that confer antibiotic resistance. This way they can be "isolated out" by growing a bunch of plasmids on a plate with a particular antibiotic. Only the plasmid vectors with the antibiotic resistance genes will survive.
- A cutting site where the plasmid can be "cut open" and the DNA fragment inserted.
There are many modern engineered plasmids which have a "polylinker" at the cutting site. A "polylinker," in a nutshell, is essentially a section of DNA which contains several cutting sites for several different restriction enzymes. This way, scientists don't have to be limited to just using EcoRI (a restriction enzyme in E. coli).
To be familiar with other cloning vectors including BACs and YACs.
BACs, or Bacterial Artificial Chromosomes, are plasmids that can be used to copy fairly large genomic fragments (100 000-300 000 base pairs). They are based on the F plasmid of E. coli (yeah, I get the feeling that we're going to be hearing a helluva lot about E. coli for the rest of my degree). Here are some of the essential characteristics in a list, because lists are great:
- It has a low copy number (i.e. few copies per cell), which apparently makes the insert more stable. Not sure how this works, though I would assume that fewer replications leads to a lower propensity for mutations.
- It contains par genes which couple plasmid replication to chromosomal replication. This ensures that every daughter cell gets at least one plasmid.
- It has resistance to chloramphenicol, so this can be used as a selectable marker. Unfortunately the plasmid without the inserted DNA also has resistance to chloramphenicol, so this can't be used to tell the recombinant DNA plasmid apart from the plain old normal plasmid. Good thing there's another built-in selectable marker- see my next point:
- The plasmid contains the lacZ gene which codes for beta-galactosidase (see my previous post on the lactose operon) AND the restriction site is located in the middle of the lacZ gene. The implications of this is that when the restriction site is cut and a fragment added, the lacZ gene ceases to function and beta-galactosidase is no longer produced. When colonies are cultured on plates containing X-gal, those colonies with a functioning lacZ gene and thus sufficient amounts of beta-galactosidase will react with X-gal to form a blue product. The recombinant DNA plasmids that do not have a functioning lacZ gene, however, will remain white.
YACs, or Yeast Artificial Chromosomes, are cloning vectors that can be used for eukaryotes. They have two TEL (telomere) sites and a CEN (centromere) site which are required for stability and cell division. YACs can exist in a circular form in bacteria. They can be turned into a linear form by digestion with BamHI- this form is required to insert the YAC into a yeast cell. As for the actual insertion process (which is actually called "transformation")- the yeast cell wall is first digested with enzymes, and then electroporation (the use of an electric current) is used to insert the YAC into the cell.
To know the important characteristics of expression vectors and how they can be used to express the protein encoded by a foreign gene.
Expression vectors are used to stimulate the production of a protein. The most important characteristic of an expression vector is that it has an inducible promoter (i.e. a promoter that you can switch on and off). For example, the lactose operon, which I've written about before, has a promoter which can be induced by allolactose (although in the lab IPTG, a lactose analogue, is more commonly used). In order to get this vector to produce something else, however, the rest of the gene is simply switched out with the gene of interest.
Subscribe to:
Posts (Atom)